


Familial Mediterranean fever is an autosomal recessive disorder characterized by recurrent bouts of fever and peritonitis, sometimes with pleuritis, erysipelas-like erythema, arthritis, and, rarely, pericarditis. Amyloidosis may develop, sometimes leading to renal failure. People with genetic origins in the Mediterranean basin are more frequently affected than other ethnic groups. Diagnosis is based on clinical suspicion and genetic testing. Treatment with prophylactic colchicine prevents acute attacks as well as amyloidosis in almost all patients. Patients who are Familial Mediterranean fever is an autosomal recessive disorder characterized by recurrent bouts of fever and peritonitis, sometimes with pleuritis, erysipelas-like erythema, arthritis, and, rarely, pericarditis. Amyloidosis may develop, sometimes leading to renal failure. People with genetic origins in the Mediterranean basin are more frequently affected than other ethnic groups. Diagnosis is based on clinical suspicion and genetic testing. Treatment with prophylactic colchicine prevents acute attacks as well as amyloidosis in almost all patients. Patients who arecolchicine-resistant or -intolerant may be treated with interleukin-1 inhibitors (anakinra, canakinumab, rilonacept). Prognosis is excellent with treatment.
Familial Mediterranean fever (FMF) is a disease of people with genetic origins in the Mediterranean basin, predominantly people who have Sephardic Jewish, North African Arab, Armenian, Turkish, Greek, or Italian ancestry. However, cases have occurred among enough other groups (eg, people who have Ashkenazi Jewish, Cuban, or Japanese ancestry), so the diagnosis should not be excluded solely on the basis of ancestry.
Etiology of Familial Mediterranean Fever
FMF is caused by
Mutations in the MEFV gene on the short arm of chromosome 16
The mutation is classically inherited in an autosomal recessive manner, but heterozygotes may manifest a clinical phenotype. FMF mutations are gain-of-function, that is, they confer new or enhanced activity on a protein, with a gene dosage effect (ie, more copies of the abnormal gene convey a greater effect). The MEFV gene normally codes a protein named pyrin, which is expressed in circulating neutrophils.
Pyrin plays a role in innate immunity. It senses modifications in the activity of the small GTPase RhoA, a molecular switch that regulates a variety of signal transduction pathways including cytoskeletal organization. Pathogen virulence toxins (such as Clostridioides difficile, Burkholderia cenocepacia, and Vibrio cholera) downregulate RhoA activity and cause an assembly pyrin along with other proteins into a pyrin inflammasome, which eventually results in the production of the proinflammatory cytokine interleukin-1 beta (IL-1 beta). MEFV pathogenic variants favor the active state of pyrin and give rise to cell membrane rupture (pyroptosis) and the release of proinflammatory cytokines (1).
There is strong evidence that Yersinia pestis, the cause of bubonic plague, led to the positive selection of FMF-associated MEFV mutations. These mutations confer a survival advantage to certain people who harbor Yersinia pestis (2).
Etiology references
1. Ben-Chetrit E: Old paradigms and new concepts in familial Mediterranean fever (FMF) - an update 2023. Rheumatology (Oxford) kead439, 2023. doi: 10.1093/rheumatology/kead439
2. Park YH, Remmers EF, Lee W, et al: Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol 21(8):857–867, 2020. doi: 10.1038/s41590-020-0705-6
Symptoms and Signs of Familial Mediterranean Fever
Onset of FMF is usually between the ages of 5 and 15 years but may be much later or earlier, even during infancy. Attacks have no regular pattern of recurrence. They usually last 12 to 72 hours but may last longer. Frequency ranges from 2 attacks/week to 1 attack/year (most commonly, once every 2 to 6 weeks). Physical and emotional stressors (eg, physical trauma, infection, menstruation) may trigger attacks (1). Severity and frequency tend to decrease during pregnancy and in patients with amyloidosis. Spontaneous remissions may last years.
Fever as high as 40° C, usually accompanied by peritonitis, is the major manifestation. Abdominal pain (usually starting in one quadrant and spreading to the whole abdomen) occurs in about 95% of patients and can vary in severity with each attack. Decreased bowel sounds, distention, guarding, and rebound tenderness are likely to occur at the peak of an attack and cannot be differentiated from a perforated viscus by physical examination. Consequently, some patients undergo urgent laparotomy before the correct diagnosis is made. With pleural involvement, dyspnea due to pleuritic pain may occur.
Other manifestations of FMF include arthritis (in 25%), usually involving the knee, ankle, and hip; an erysipelas-like rash of the lower leg; and scrotal swelling and pain caused by inflammation of the tunica vaginalis of the testis. Pericarditis occurs rarely. The pleural, synovial, and skin manifestations of FMF vary in frequency among different populations (2).
Despite the severity of symptoms during acute attacks, most patients recover swiftly and remain free of illness until their next attack.
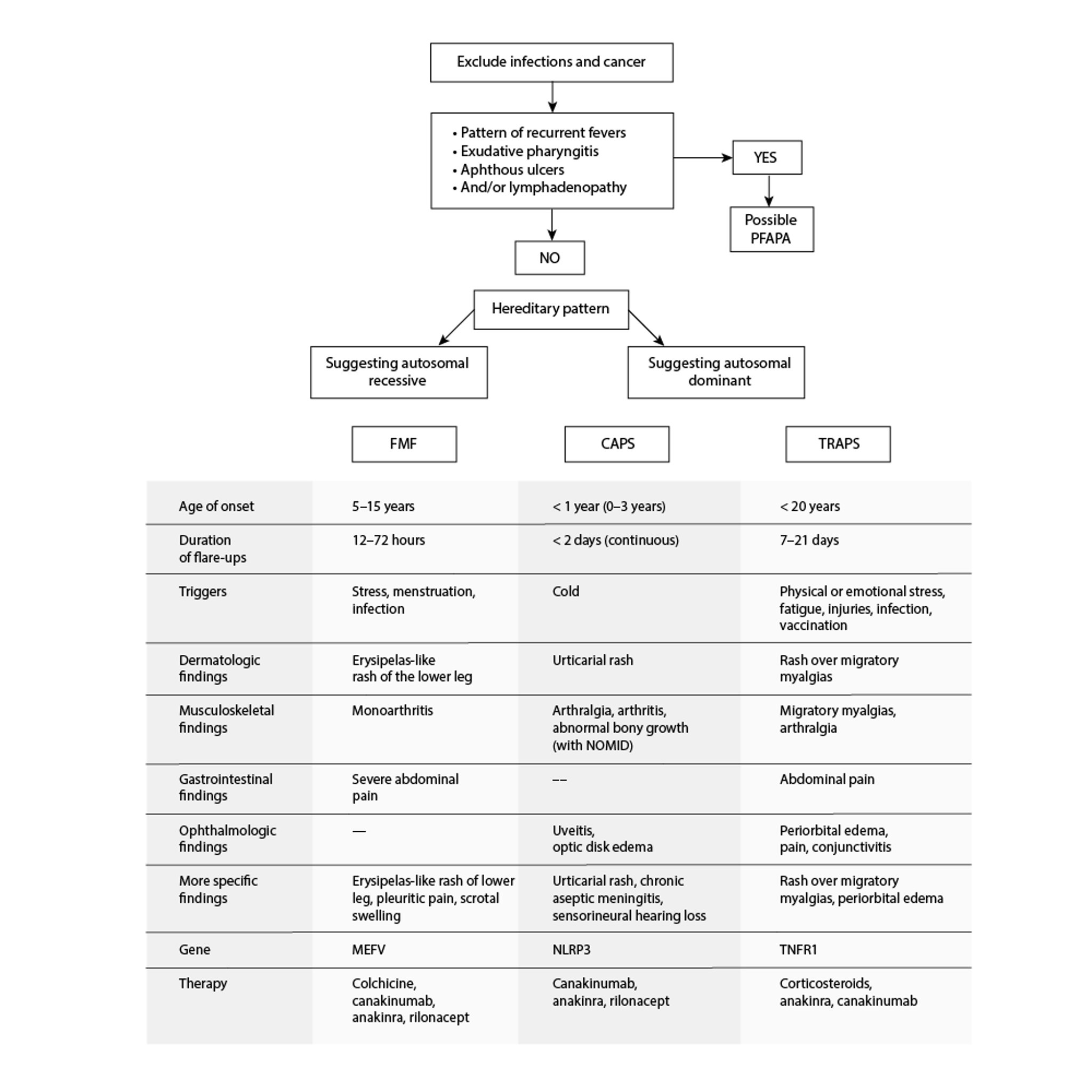
Autoinflammatory Periodic Fever Disorders
CAPS = cryopyrin-associated periodic syndromes; FMF = familial Mediterranean fever; NOMID = neonatal-onset multisystem inflammatory disease; PFAPA = periodic fevers with aphthous stomatitis, pharyngitis, and adenitis; TRAPS = tumor necrosis factor receptor–associated periodic syndrome. Adapted from Sag E, Bilginer Y, Ozen S: Autoinflammatory diseases with periodic fevers. Curr Rheumatol Rep 19(7):41, 2017. doi: 10.1007/s11926-017-0670-8 |

Complications of familial Mediterranean fever
The most significant long-term complication of FMF is
Chronic renal failure caused by deposition of amyloid protein in the kidneys
Amyloid may also be deposited in the gastrointestinal tract, liver, spleen, heart, testes, and thyroid.
FMF may cause infertility or spontaneous abortion in up to one third of untreated women because peritoneal pelvic adhesions form, interfering with conception.
FMF increases the risk of other inflammatory disorders, such as ankylosing spondylitis, immunoglobulin A–associated (IgA) vasculitis, polyarteritis nodosa, and Behçet disease (3).
Symptoms and signs references
1. Yenokyan G, Armenian HK: Triggers for attacks in familial Mediterranean fever: application of the case-crossover design. Am J Epidemiol 175(10):1054-1061, 2012. doi: 10.1093/aje/kwr460
2. Ben-Chetrit E, Yazici H: Familial Mediterranean fever: different faces around the world. Clin Exp Rheumatol 37 Suppl 121(6):18-22, 2019. PMID: 31694745
3. Balcı-Peynircioğlu B, Kaya-Akça Ü, Arıcı ZS, et al: Comorbidities in familial Mediterranean fever: Analysis of 2000 genetically confirmed patients. Rheumatology (Oxford) 59(6):1372–1380, 2020. doi: 10.1093/rheumatology/kez410
Diagnosis of Familial Mediterranean Fever
Clinical evaluation
Genetic testing
Diagnosis of familial Mediterranean fever is mainly clinical based on Tel HaShomer criteria (see table Tel HaShomer Criteria for the Diagnosis of Familial Mediterranean Fever) (1), but genetic testing is available and is particularly useful in evaluation of atypical cases. However, current genetic testing is not infallible; some patients with phenotypically unmistakeable FMF have only a single mutated gene or occasionally no evident mutations in the MEFV gene. About 10 to 20% of patients who meet the diagnostic criteria for FMF do not have MEFV mutations, which suggests epigenetic and environmental factors contribute to the disease pathogenesis (2).
Nonspecific findings include elevations in white blood cells with neutrophil predominance, erythrocyte sedimentation rate, C-reactive protein, and fibrinogen. Urinary excretion of > 0.5 g protein/24 hours may suggest renal amyloidosis.
Differential diagnosis includes acute intermittent porphyria, hereditary angioedema with abdominal attacks, relapsing pancreatitis, and other hereditary relapsing fevers.
Tel HaShomer Criteria for the Diagnosis of Familial Mediterranean Fever*
Major Criteria |
Typical attacks† of
|
Incomplete attacks‡ of the abdomen |
Minor Criteria |
Incomplete attacks‡ involving the chest, joints, or both |
Leg pain with exertion |
Favorable response to treatment with colchicineFavorable response to treatment with colchicine |
* Diagnosis requires ≥ 1 major criterion or ≥ 2 minor criteria. |
† Typical attacks are recurrent (at least 3 episodes of the same type), febrile (rectal temperature ≥ 38° C), and short in duration (12 to 72 hours). |
‡ Incomplete attacks are painful and recurrent. They differ from typical attacks in 1 or 2 features:
|
Data from Livneh A, Langevitz P, Zemer D, et al: Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40(10):1879–1885, 1997. doi: 10.1002/art.1780401023 |
Diagnosis references
1. Livneh A, Langevitz P, Zemer D, et al: Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40(10):1879–1885, 1997. doi: 10.1002/art.1780401023
2. Booty MG, Chae JJ, Masters SL, et al: Familial Mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum 60(6):1851–1861, 2009. doi: 10.1002/art.24569
Treatment of Familial Mediterranean Fever
Daily colchicine
In patients who are colchicine-resistant or -intolerant, interleukin-1 (IL-1) inhibitors
Daily prophylactic colchicine should be initiated as soon as the diagnosis is made to prevent attacks as well as Daily prophylactic colchicine should be initiated as soon as the diagnosis is made to prevent attacks as well asamyloidosis (1). Colchicine provides complete remission or distinct improvement in almost 95% of patients. If attacks or subclinical inflammation persist, the colchicine dose should be increased. Initiation of colchicine at the peak of an attack is not beneficial. Children require a dose adjustment for colchicine that is typically based on age, weight, and severity of phenotype and genotype (2). Dose adjustments should also be made for patients with liver or kidney dysfunction. Widespread use of prophylactic colchicine has led to a dramatic reduction in the incidence of amyloidosis and subsequent renal failure.). Dose adjustments should also be made for patients with liver or kidney dysfunction. Widespread use of prophylactic colchicine has led to a dramatic reduction in the incidence of amyloidosis and subsequent renal failure.
Colchicine does not add to the increased risk of infertility or spontaneous abortion among affected women; when taken during pregnancy, it does not increase the risk of teratogenic events. Lack of response to Colchicine does not add to the increased risk of infertility or spontaneous abortion among affected women; when taken during pregnancy, it does not increase the risk of teratogenic events. Lack of response tocolchicine is often caused by poor adherence to the regimen.
Patients who are colchicine-resistant or -intolerant may be treated with IL-1 inhibitors (anakinra once a day, rilonacept weekly, or canakinumab every 4 weeks) (-resistant or -intolerant may be treated with IL-1 inhibitors (anakinra once a day, rilonacept weekly, or canakinumab every 4 weeks) (3, 4). However, the role of IL-1 inhibitors in preventing amyloidosis remains unknown, and patients taking IL-inhibitors should continue taking colchicine if tolerated.). However, the role of IL-1 inhibitors in preventing amyloidosis remains unknown, and patients taking IL-inhibitors should continue taking colchicine if tolerated.
Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen are sometimes needed for analgesia.Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen are sometimes needed for analgesia.
Treatment references
1. Ter Haar N, Lachmann H, Özen S, et al: Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis 72(5):678-685, 2013. doi: 10.1136/annrheumdis-2011-201268
2. Goldberg O, Levinsky Y, Peled O, et al: Age dependent safety and efficacy of colchicine treatment for familial mediterranean fever in children. : Age dependent safety and efficacy of colchicine treatment for familial mediterranean fever in children.Semin Arthritis Rheum 49(3):459-463, 2019. doi: 10.1016/j.semarthrit.2019.05.011
2. Ozen S, Demirkaya E, Erer B, et al: EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis 75(4):644–651, 2016. doi: 10.1136/annrheumdis-2015-208690
3. De Benedetti F, Gattorno M, Anton J, et al: Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 378(20):1908–1919, 2018. doi: 10.1056/NEJMoa1706314
Key Points
Familial Mediterranean fever (FMF) is caused by an autosomal recessive mutation in the MEFV gene, which encodes the pyrin protein that helps modulate the inflammatory response in neutrophils.
People with genetic origins in the Mediterranean basin are more commonly (but not exclusively) affected.
Patients have brief episodes of fever, abdominal pain, and sometimes other symptoms such as pleuritis, arthritis, and rash.
Renal amyloidosis, sometimes causing renal failure, is the most common complication, but prophylactic colchicine provides protection against amyloidosis.
Diagnose clinically; genetic testing is available and may support the diagnosis.
Daily colchicine results in significant protection against attacks in most patients, but patients who are Daily colchicine results in significant protection against attacks in most patients, but patients who arecolchicine-resistant or -intolerant may be given an IL-1 inhibitor (anakinra, rilonacept, or canakinumab).-resistant or -intolerant may be given an IL-1 inhibitor (anakinra, rilonacept, or canakinumab).







