



- Introduction to Inherited Disorders of Metabolism
- Approach to the Patient With a Suspected Inherited Disorder of Metabolism
- Mitochondrial Oxidative Phosphorylation Disorders
- Peroxisomal Disorders
- Overview of Amino Acid and Organic Acid Metabolism Disorders
- Branched-Chain Amino Acid Metabolism Disorders
- Hartnup Disease
- Methionine Metabolism Disorders
- Phenylketonuria (PKU)
- Tyrosine Metabolism Disorders
- Urea Cycle Disorders
- Overview of Carbohydrate Metabolism Disorders
- Fructose Metabolism Disorders
- Galactosemia
- Glycogen Storage Diseases
- Pyruvate Metabolism Disorders
- Other Carbohydrate Metabolism Disorders
- Overview of Fatty Acid and Glycerol Metabolism Disorders
- Beta-Oxidation Cycle Disorders
- Glycerol Metabolism Disorders
- Overview of Lysosomal Storage Disorders
- Cholesteryl Ester Storage Disease and Wolman Disease
- Fabry Disease
- Gaucher Disease
- Krabbe Disease
- Metachromatic Leukodystrophy
- Niemann-Pick Disease
- Tay-Sachs Disease and Sandhoff Disease
- Overview of Purine and Pyrimidine Metabolism Disorders
- Purine Catabolism Disorders
- Purine Nucleotide Synthesis Disorders
- Purine Salvage Disorders
- Pyrimidine Metabolism Disorders
Topic Resources

Gangliosides are complex sphingolipids present in the brain. There are 2 major forms, GM1 and GM2, both of which may be involved in lysosomal storage disorders. There are 2 main types of GM2 gangliosidosis, each of which can be caused by numerous different mutations.
For more information, see table Some Sphingolipidoses.
See also Approach to the Patient With a Suspected Inherited Disorder of Metabolism.
Tay-Sachs Disease
Deficiency of hexosaminidase A results in accumulation of GM2 in the brain. Inheritance is autosomal recessive; the most common mutations are carried by 1/27 unaffected adults of Eastern European (Ashkenazi) Jewish origin, although other mutations cluster in some French-Canadian and Cajun populations. The disease develops in 25% of the children when both parents are carriers.
Children with Tay-Sachs disease start missing developmental milestones after age 6 months and develop progressive cognitive and motor deterioration resulting in seizures, intellectual disability, paralysis, and death by age 5 years.
A cherry-red macular spot is common.

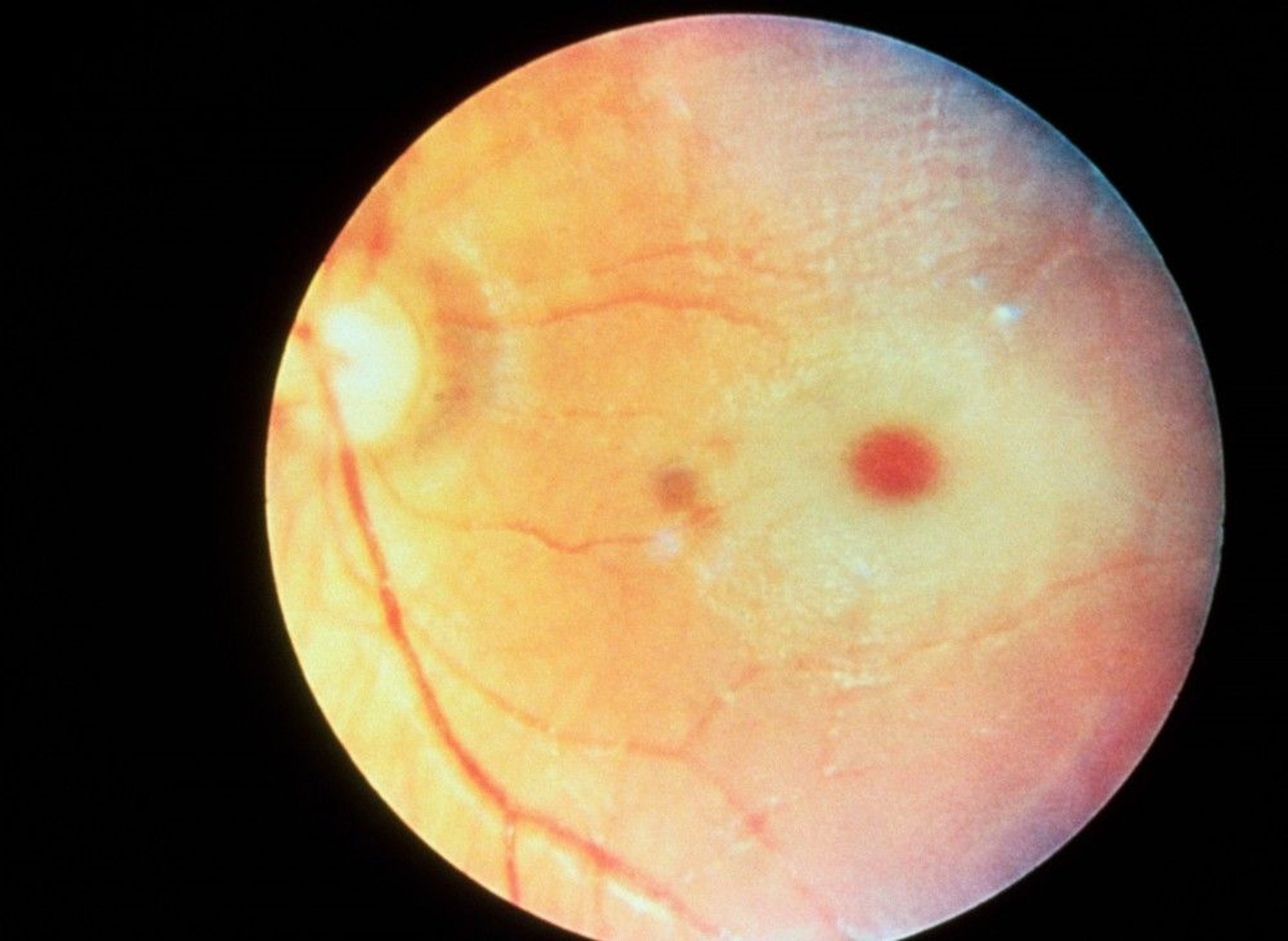
RALPH C. EAGLE, JR./SCIENCE PHOTO LIBRARY
Diagnosis of Tay-Sachs disease is clinical and can be confirmed by DNA analysis and/or enzyme assay. (See also testing for suspected inherited disorders of metabolism.)
In the absence of effective treatment, management is focused on screening adults of childbearing age in high-risk populations to identify carriers (by way of enzyme activity and mutation testing) combined with genetic counseling.
Sandhoff Disease
There is a combined hexosaminidase A and B deficiency. Clinical manifestations include progressive cerebral degeneration beginning at 6 months, accompanied by blindness, cherry-red macular spot, and hyperacusis.
Sandhoff disease is almost indistinguishable from Tay-Sachs disease in course, diagnosis, and management, except that there is visceral involvement (hepatomegaly and bone change) and no ethnic association.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Online Mendelian Inheritance in Man (OMIM) database: Complete gene, molecular, and chromosomal location information






