


Osteosclerosis is a type of osteopetrosis that involves abnormal hardening of bone and increased skeletal density with little disturbance of modeling; in some types, bony encroachment on the marrow cavity causes cytopenias.
")
Osteopetrosis with delayed manifestations (Albers-Schönberg disease)
This type of osteopetrosis is autosomal dominant, manifesting during childhood, adolescence, or young adulthood. It is less severe than osteopetrosis with precocious manifestations. The defective CLCN7 gene encodes a chloride channel that is apparently important in osteoclast function.
Osteopetrosis with delayed manifestations is relatively common and has a wide geographic and ethnic distribution. Affected people may be asymptomatic; general health is usually unimpaired. However, facial palsy and deafness may occur due to cranial nerve entrapment. Bony overgrowths may narrow the marrow cavity and cause cytopenias ranging from anemia to pancytopenia. Extramedullary hematopoiesis may occur, resulting in hepatosplenomegaly; consequent hypersplenism may worsen anemia.
The skeleton usually is radiologically normal at birth. However, bone sclerosis becomes increasingly apparent as children age, and diagnosis is typically based on x-rays done for unrelated reasons. Bony involvement is widespread but patchy. The calvaria is dense, and sinuses may be obliterated. Sclerosis of the vertebral end plate causes the characteristic rugby-shirt appearance (horizontal banding).
Some patients require transfusion or splenectomy to treat anemia.
Osteopetrosis with precocious manifestations
This type of osteopetrosis is autosomal recessive, malignant, and congenital, manifesting during infancy. It is uncommon, frequently lethal, and often due to a mutation in the osteoclast-associated gene TCIRG1. Bony overgrowth progressively obliterates the marrow cavity, causing severe pancytopenia.
Initial symptoms of osteopetrosis with precocious manifestations include failure to thrive, spontaneous bruising, abnormal bleeding, and anemia. Palsies of the 2nd, 3rd, and 7th cranial nerves and hepatosplenomegaly occur later. Bone marrow failure (anemia, overwhelming infection, or hemorrhage) usually causes death in the first year of life.
Diagnosis of osteopetrosis with precocious manifestations is suspected by the presence of bony overgrowths in the context of anemia, unusual bleeding, and poor growth. Typically, plain x-rays are done, along with complete blood count and coagulation tests. General increased bone density is the predominant feature on x-ray. Penetrated x-rays of long bones show transverse bands in the metaphyseal regions and longitudinal striations in the shafts. As the disorder progresses, the ends of the long bones, particularly the proximal humerus and distal femur, become flask-shaped. Characteristic endobones (bone within a bone) form in the vertebrae, pelvis, and tubular bones. The skull becomes thickened, and the spine has a rugby-shirt appearance.
Hematopoietic stem cell transplantation has had excellent results. However, prognosis is poor with human leukocyte antigen (HLA)–mismatched grafts. Prednisone, calcitriol, and interferon gamma are effective in some cases.has had excellent results. However, prognosis is poor with human leukocyte antigen (HLA)–mismatched grafts. Prednisone, calcitriol, and interferon gamma are effective in some cases.
Osteopetrosis with renal tubular acidosis
This type of osteopetrosis is autosomal recessive. The genetic defect involves mutations of the gene encoding carbonic anhydrase II.
Osteopetrosis with renal tubular acidosis causes weakness, stunted stature, and failure to thrive.
Bones appear dense on x-rays, and cerebral calcifications are seen; renal tubular acidosis is present, and red blood cell carbonic anhydrase activity is decreased.
Hematopoietic stem cell transplantation cures the osteopetrosis but has no effect on the renal tubular acidosis. Maintenance therapy consists of bicarbonate and electrolyte supplementation to correct renal losses.
Pyknodysostosis
This autosomal recessive disorder is caused by loss of function mutations in the gene encoding cathepsin K, an osteoclast-derived protease important in degradation of extracellular bone matrix.
Short stature becomes evident in early childhood; adult height is ≤ 150 cm (5 ft). Other manifestations include an enlarged skull, short and broad hands and feet, short sclerotic terminal phalanges, dystrophic nails, and retention of primary teeth. Blue sclerae (due to a deficiency in connective tissue allowing the underlying vessels to show through) are usually recognized during infancy. Affected people resemble each other closely; they have a small face, a receding chin, and carious, misplaced teeth. The cranium bulges, and the anterior fontanelle remains patent. Pathologic fractures are a complication of pyknodysostosis.

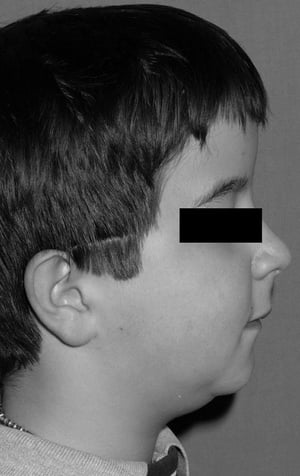
This photo shows a receding chin in a boy with pyknodysostosis.
© Springer Science+Business Media


This photo shows short, broad hands and dysplasia of terminal phalanges (particularly of the thumbs) in a man with pyknodysostosis.
© Springer Science+Business Media

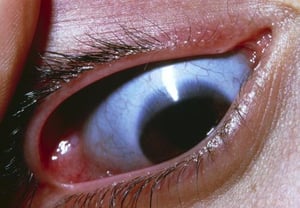
This photo shows a close-up of the eye showing a blue sclera (normally white).
JAMES STEVENSON/SCIENCE PHOTO LIBRARY
This photo shows a receding chin in a boy with pyknodysostosis.
© Springer Science+Business Media
This photo shows short, broad hands and dysplasia of terminal phalanges (particularly of the thumbs) in a man with pyknodysostosis.
© Springer Science+Business Media
This photo shows a close-up of the eye showing a blue sclera (normally white).
JAMES STEVENSON/SCIENCE PHOTO LIBRARY
Diagnosis of pyknodysostosis is suspected by the presence of blue sclerae, short stature, and characteristic skeletal features. Typically, plain x-rays are done. Bone sclerosis appears on x-rays during childhood, but neither bone striations nor endobones (bone within bone) are seen. Facial bones and paranasal sinuses are hypoplastic, and the mandibular angle is obtuse. Clavicles may be gracile, and their lateral portions may be underdeveloped; distal phalanges are rudimentary.
Plastic surgery has been used to correct severe deformities of the face and jaw.







