


Hirschsprung disease is a congenital anomaly consisting of a failure of neuronal colonization (and thus a failure of innervation) of the lower intestine, usually limited to the colon, resulting in partial or total functional obstruction. Symptoms are obstipation and distention. The diagnosis may be suggested by evaluation with a barium enema; definitive diagnosis requires a rectal biopsy (suction biopsy or surgical biopsy). Anal manometry can help in the evaluation and reveals lack of relaxation of the internal anal sphincter upon rectal balloon distention. Treatment is surgical.

(Also see Overview of Congenital Gastrointestinal Anomalies.)
Hirschsprung disease is caused by congenital absence of the Meissner and Auerbach autonomic plexus (aganglionosis) in the intestinal wall. The estimated incidence is 1 in 5000 live births (1). Disease is usually limited to the distal colon (75% of cases) but can involve the entire colon (3 to 10% of cases) or even the entire large and small bowels; the denervated area is always contiguous (2). Males are more commonly affected (male:female ratio 4:1) unless the entire colon is involved, in which case there is no gender difference.

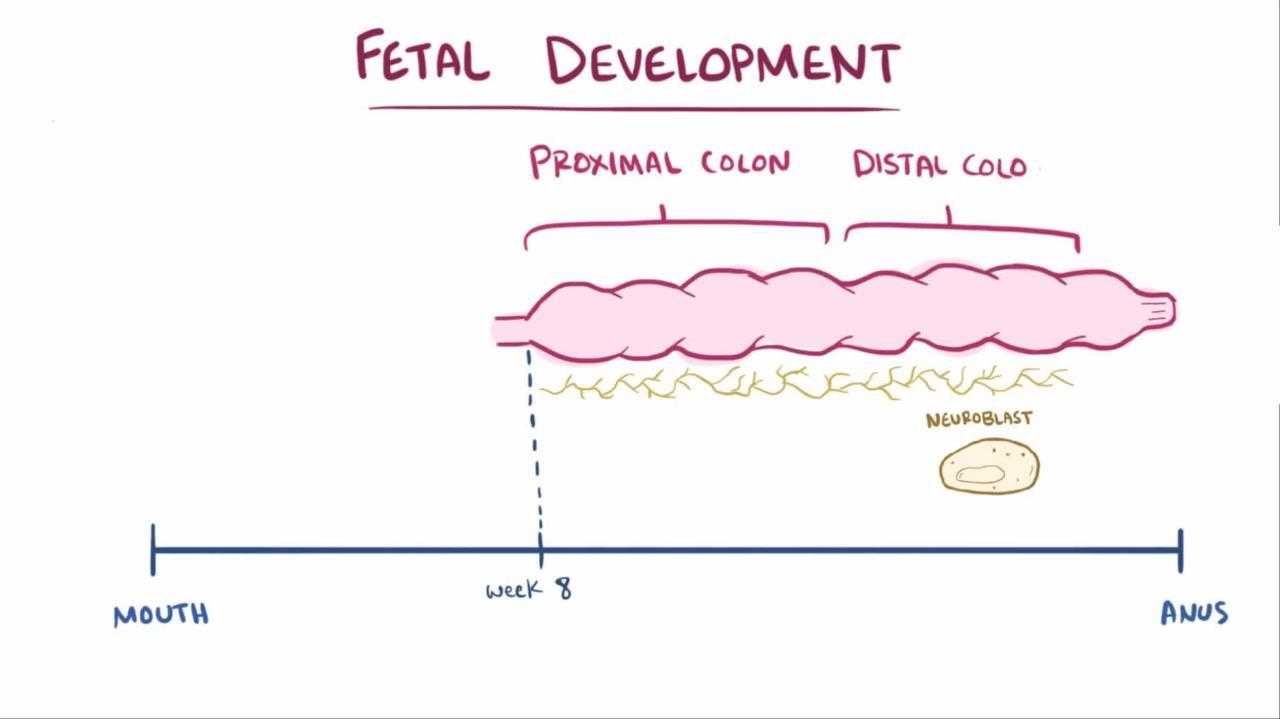
The etiology of the aganglionosis is thought to be the failure of migration of neural progenitors from the neural crest. There is a significant genetic component to this disorder, and at least 12 different genetic mutations are associated with Hirschsprung. The likelihood of disease among family members increases with increasing length of the involved gut—3 to 8% for disease of the distal colon and up to 20% for disease involving the entire colon. Twenty to 25% of patients with Hirschsprung disease have another congenital anomaly, and 15% have a genetic abnormality (Down syndrome is the most common). Approximately 20% of patients with congenital central hypoventilation syndrome also have Hirschsprung disease; the combination is referred to as Haddad syndrome. Other disorders associated with Hirschsprung disease include Waardenburg syndrome, Bardet-Biedl syndrome, Goldberg-Shprintzen syndrome, and cartilage-hair hypoplasia.
Pearls & Pitfalls
|
Peristalsis in the involved segment is absent or abnormal, resulting in continuous smooth muscle spasm and partial or complete obstruction with accumulation of intestinal contents and massive dilation of the more proximal, normally innervated intestine. Skip lesions almost never occur.
General references
1. Mueller JL, Goldstein AM: The science of Hirschsprung disease: What we know and where we are headed. Semin Pediatr Surg 31(2):151157, 2022. doi:10.1016/j.sempedsurg.2022.151157
2. Urla C, Lieber J, Obermayr F, et al: Surgical treatment of children with total colonic aganglionosis: functional and metabolic long-term outcome. BMC Surg 18(1):58, 2018. Published 2018 Aug 15. doi:10.1186/s12893-018-0383-6
Symptoms and Signs of Hirschsprung Disease
Patients most commonly present early in life, but some do not present until childhood or even adulthood.
Normally, almost all neonates pass meconium in the first 24 hours of life. Approximately 50 to 90% of neonates with Hirschsprung disease fail to pass meconium in the first 48 hours of life. Infants present with obstipation, abdominal distention, and, finally, vomiting as in other forms of distal bowel obstruction. Occasionally, infants with ultrashort segment aganglionosis have only mild or intermittent constipation, often with intervening bouts of mild diarrhea, resulting in delay in diagnosis.
In older infants and children, symptoms and signs may include anorexia, constipation, lack of a physiologic urge to defecate, and, on digital rectal examination, an empty rectum with stool palpable higher up in the colon and an explosive passage of stool upon withdrawal of the examining finger (blast sign). Infants may also fail to thrive. Less commonly, infants may present with Hirschsprung enterocolitis.
Diagnosis of Hirschsprung Disease
Barium enema
Rectal suction or surgical biopsy
Rectal manometry
Diagnosis of Hirschsprung disease should be made as soon as possible. The longer the disease goes untreated, the greater the chance of developing Hirschsprung enterocolitis, a complication that may be fulminant and fatal. Most patients can be diagnosed in early infancy.

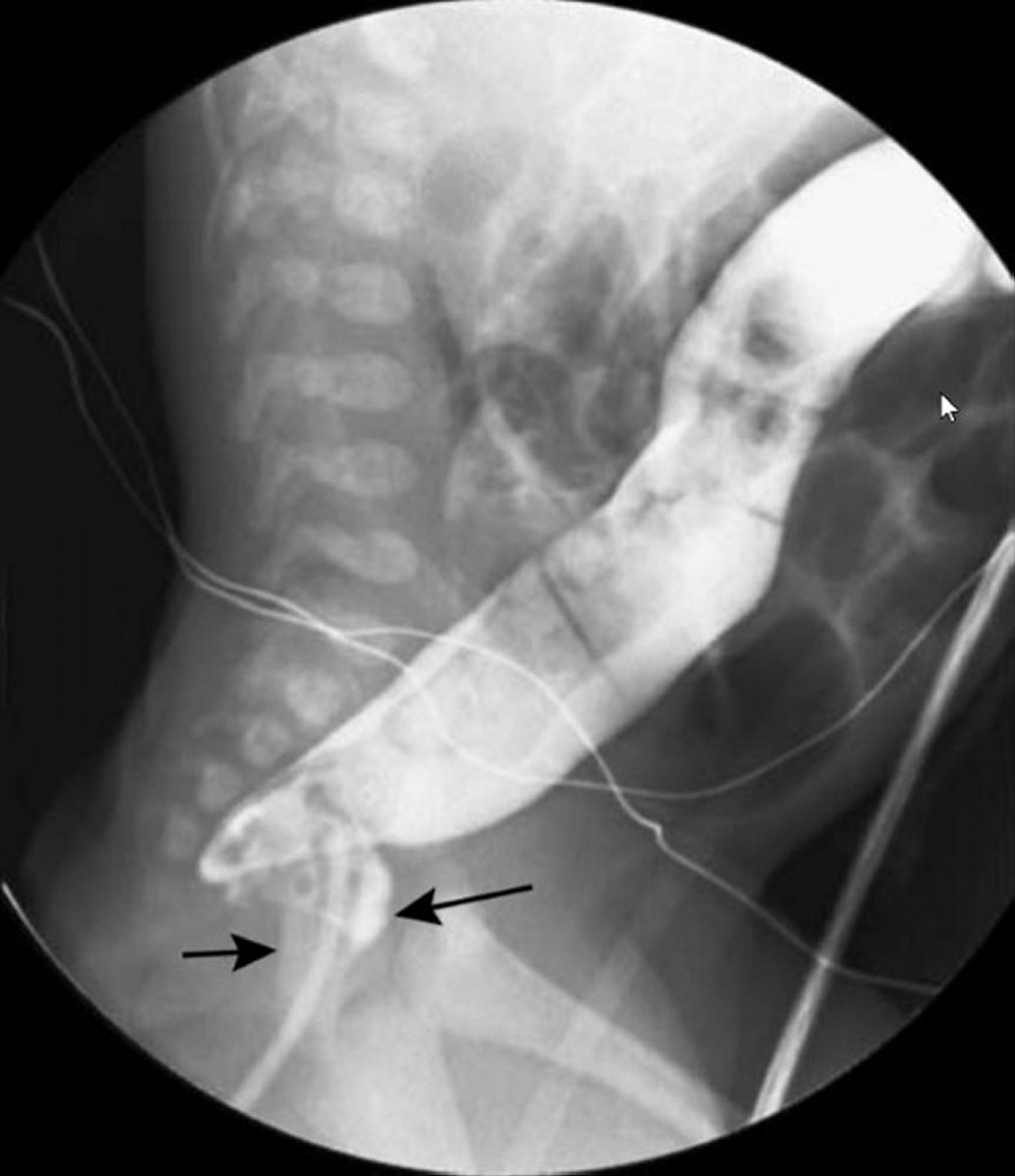
© Springer Science+Business Media
Initial approach is typically with barium enema and/or rectal suction biopsy. (A barium enema should not be done in patients suspected of having Hirschsprung enterocolitis because of the risk of perforation.) Barium enema may show a transition in diameter between the dilated, normally innervated colon proximal to the narrowed distal segment (which lacks normal innervation). Barium enema should be done without prior preparation, which can dilate the abnormal segment and decompress the proximal colon, rendering the test nondiagnostic. Because characteristic findings may not be present in the neonatal period, a 24-hour post-evacuation x-ray should be taken; if the colon is still filled with barium, Hirschsprung disease is likely.
A rectal suction biopsy can disclose the absence of ganglion cells. Acetylcholinesterase staining can be done to highlight the enlarged nerve trunks; calretinin staining can be done to detect the absence of mucosal innervation (1).
Some centers also can do rectal manometry, which typically reveals a lack of relaxation of the internal anal sphincter upon balloon insufflation of the rectum, which is characteristic of the abnormal innervation.
Definitive diagnosis requires a surgical or suction biopsy of the rectum and then surgical biopsies to map the extent of disease and thus plan surgical treatment.
Genetic testing is not routine but may be done if evaluation shows manifestations of a genetic syndrome or in cases of total colonic aganglionosis.


Pearls & Pitfalls
|
Diagnosis reference
1. Guinard-Samuel V, Bonnard A, De Lagausie P, et al: Calretinin immunohistochemistry: a simple and efficient tool to diagnose Hirschsprung disease. Mod Pathol 22(10):1379-1384, 2009. doi:10.1038/modpathol.2009.110
Treatment of Hirschsprung Disease
Surgical repair
Treatment of Hirschsprung disease is surgical repair by bringing normally innervated bowel to the anus with preservation of the anal sphincters. In the neonate, this had typically been a 2-stage procedure beginning with a colostomy proximal to the aganglionic segment to decompress the colon. Then the neonate was allowed to grow before the 2nd stage of the procedure, in which the entire aganglionic portion of the colon was resected and a pull-through procedure was done. However, many centers now do a 1-stage procedure in the neonatal period for short-segment disease. Results using laparoscopic technique are similar to those of the open method and are associated with shorter hospitalizations, earlier initiation of feeding, and less pain.
After definitive repair, the prognosis is good, although a number of infants have chronic dysmotility with constipation, obstructive problems, or both.
Key Points
Congenital denervation affects the distal colon and less often larger regions of the colon and sometimes even the small bowel.
Infants typically present with findings of distal bowel obstruction, such as obstipation, abdominal distention, and vomiting.
Barium enema findings (done without prior preparation) and rectal manometry are highly suggestive; diagnosis is confirmed by rectal biopsy.
The affected segment is resected surgically.
Hirschsprung Enterocolitis (Toxic Megacolon)
Hirschsprung enterocolitis is a life-threatening complication of Hirschsprung disease resulting in a grossly enlarged colon, often followed by sepsis and shock.
The etiology of Hirschsprung enterocolitis seems to be marked proximal dilation secondary to obstruction, with thinning of the colonic wall, bacterial overgrowth, and translocation of gut bacteria. Sepsis or shock can develop (more often when the entire colon is affected by Hirschsprung), and death can follow rapidly; mortality rate is about 1%. Close monitoring of infants with Hirschsprung disease is therefore essential.
Hirschsprung enterocolitis occurs most commonly in the first several months of life before surgical correction but can occur postoperatively, typically in the first year after surgery. Infants with Down syndrome are at increased risk of developing this complication postoperatively. Infants present with fever, abdominal distention, diarrhea (which may be bloody), and, subsequently, obstipation.
A barium enema should not be done in patients suspected of having Hirschsprung enterocolitis because of the risk of perforation.
Initial treatment of Hirschsprung enterocolitis is supportive with fluid resuscitation, decompression with a nasogastric and rectal tube, and broad-spectrum antibiotics to include anaerobic coverage (eg, a combination of ampicillin, gentamicin, and metronidazole; meropenem alone; or piperacillin/tazobactam). Some experts advocate saline enemas in infants who are not critically ill to clean out the colon, but this must be done carefully so as not to increase colonic pressure and cause perforation. Initial treatment of Hirschsprung enterocolitis is supportive with fluid resuscitation, decompression with a nasogastric and rectal tube, and broad-spectrum antibiotics to include anaerobic coverage (eg, a combination of ampicillin, gentamicin, and metronidazole; meropenem alone; or piperacillin/tazobactam). Some experts advocate saline enemas in infants who are not critically ill to clean out the colon, but this must be done carefully so as not to increase colonic pressure and cause perforation.
Surgery is the definitive treatment for infants who have not yet undergone surgical repair, as well as for those with perforation or necrotic gut. For infants who can be stabilized, definitive surgical therapy can be done instead of the previous standard approach of a diverting ileostomy or colostomy. The previous standard approach should be done in those with severe disease.







