


La esclerosis lateral amiotrófica (enfermedad Lou Gehrig) es la forma más habitual de enfermedad de la motoneurona.
Por lo general, los músculos se debilitan y se atrofian, y los movimientos se vuelven rígidos, torpes y progresivamente más difíciles de ejecutar.
El diagnóstico se basa sobre todo en los resultados de la exploración física, y realiza la electromiografía, los estudios de conducción nerviosa, la resonancia magnética nuclear y los análisis de sangre para contribuir a confirmar el diagnóstico.
Aunque los fármacos ayudan a disminuir los síntomas, no existe cura específicos.
(Véase también Introducción al sistema nervioso periférico.)
Las enfermedades de las neuronas motoras pueden afectar el sistema nervioso central (cerebro y médula espinal), así como el sistema nervioso periférico (nervios fuera del encéfalo y la médula espinal).
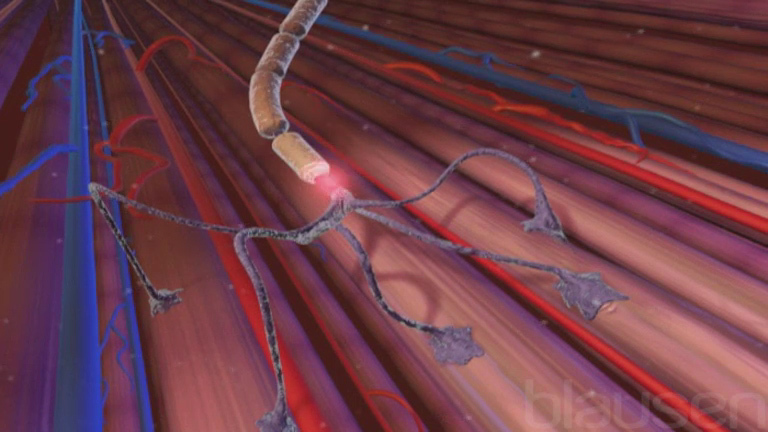
Para que exista una función muscular normal, el tejido muscular y las conexiones nerviosas entre el cerebro y los músculos deben ser normales. El movimiento de los músculos es iniciado por las células nerviosas (neuronas) que se encuentran en la médula espinal y en la parte frontal del cerebro (denominada corteza motora). Las células nerviosas de la corteza motora se conectan con las células nerviosas de la médula espinal que estimulan el movimiento de los músculos (llamados nervios motores). En las enfermedades de las neuronas motoras, estas células nerviosas se erosionan progresivamente y los nervios periféricos que las conectan con el músculo se deterioran. Como consecuencia, los músculos se debilitan, se desgastan (atrofian) y se paralizan por completo incluso aunque los propios músculos no sean la causa del problema.
Usar el cerebro para mover un músculo
Mover un músculo por lo general implica la comunicación entre el músculo y el cerebro a través de los nervios. El ímpetu para mover un músculo puede originarse en el cerebro, como cuando una persona decide conscientemente mover un músculo, por ejemplo, para levantar un libro. O el impulso de mover un músculo tiene su origen en los sentidos. Por ejemplo, las terminaciones nerviosas especiales de la piel (receptores sensoriales) permiten sentir el dolor o un cambio de temperatura. Esta información sensorial se envía al cerebro y este envía un mensaje al músculo para saber cómo responder. En este tipo de intercambio intervienen dos vías nerviosas complejas:
| |
| |
Si la sensación se produce de repente y es grave (como sucede al pisar una piedra afilada o al tomar una taza de café muy caliente), el impulso puede viajar a la médula espinal y directamente de vuelta al nervio motor, sin pasar por el cerebro. El resultado es una respuesta rápida de un músculo, al retirarse inmediatamente de lo que sea que esté causando el dolor. Esta respuesta se denomina reflejo vertebral. | |


Las enfermedades de las neuronas motoras presentan varias formas, como las siguientes:
Esclerosis lateral amiotrófica (los más comunes)
Las enfermedades de las neuronas motoras son más frecuentes entre los varones y suelen aparecer entre los 50 y los 70 años de edad. Por lo general, se desconoce la causa. Algunas personas que presentan una enfermedad de las neuronas motoras tienen un tipo hereditario (causado por una mutación en un gen), y por tanto algunos de sus familiares también tienen la enfermedad.
Diferentes partes del sistema nervioso pueden verse afectadas en primer lugar. Por ejemplo, algunas formas de enfermedad de la motoneurona afectan inicialmente a la boca y la garganta. Otras afectan a una mano o un pie primero o les afectan más severamente.
La parálisis de larga duración puede conducir a un acortamiento permanente de los músculos (contracturas).
Síntomas de la ELA y de otras enfermedades de las neuronas motoras
Aunque no se sienta dolor y no exista alteración alguna de la sensibilidad, la fuerza muscular está afectada. La depresión es frecuente.
Esclerosis lateral amiotrófica (enfermedad Lou Gehrig)
La enfermedad de la motoneurona progresiva comienza con debilidad, a menudo en las manos y con menos frecuencia en los pies o en la boca y la garganta. La debilidad progresa más en un lado del cuerpo que en el otro, y suele ir ascendiendo por el brazo o por la pierna. Los músculos, por lo general los que están en las manos y pies, comienzan a disminuir de volumen (atrofia). Los calambres musculares también son frecuentes y aparecen antes que la debilidad, pero no se producen alteraciones en la sensibilidad. Las personas afectadas pierden peso y se sienten inexplicablemente cansadas.
Con el paso del tiempo, aumenta la debilidad.
Cuando la esclerosis lateral amiotrófica afecta los nervios motores del cerebro, el tono muscular suele aumentar y los músculos tienden a endurecerse y tensarse, dando lugar a espasmos musculares (lo que se denomina espasticidad). Los movimientos son rígidos y torpes. Cuando los nervios motores de la médula espinal se ven afectados, el tono muscular disminuye, lo que hace que las extremidades parezcan flojas y flácidas. Cuando se pierde la conexión entre los nervios motores y los músculos, los músculos se contraen de forma espontánea (lo que se denomina fasciculaciones).
A veces hay dificultad para controlar las expresiones faciales. La debilidad de los músculos de la garganta provoca una mala articulación del lenguaje y dificultad para deglutir (disfagia). Debido a que la deglución es difícil, el babeo y el ahogo al ingerir líquidos son habituales. Los alimentos o la saliva pueden ser inhalados (aspiración) hacia el interior de los pulmones, lo que aumenta el riesgo de neumonía (denominada neumonía por aspiración). La voz normalmente suena nasal, pero puede ser ronca.
A medida que los síntomas progresan las personas son incapaces de controlar sus respuestas emocionales, y ríen o lloran sin motivo.
Finalmente, los músculos implicados en la respiración se debilitan, lo que conlleva la aparición de problemas respiratorios. Algunas personas necesitan una máquina de respiración artificial para respirar.
La rapidez con que avanza la esclerosis lateral amiotrófica es variable:
Alrededor del 50% de las personas que sufren este trastorno mueren en un periodo de 3 años desde que aparecen los primeros síntomas.
Alrededor del 20% viven 5 años.
Alrededor del 10% viven 10 años o más.
Unas cuantas personas sobreviven más de 30 años.
Esclerosis lateral primaria y parálisis seudobulbar progresiva
Estas enfermedades de las neuronas motoras son variantes de la esclerosis lateral amiotrófica muy poco frecuentes:
La esclerosis lateral primaria afecta principalmente los brazos y las piernas.
La parálisis seudobulbar progresiva afecta sobre todo los músculos de la cara, la mandíbula y la garganta.
En ambos trastornos, los músculos están débiles y muy rígidos y tensos (espásticos), pero no se producen espasmos musculares (lo que se denomina fasciculaciones) ni atrofia.
Labilidad emocional: las personas con parálisis seudobulbar progresiva pueden pasar rápidamente de la alegría a la tristeza sin un motivo aparente. Los arrebatos emocionales inapropiados (por ejemplo, risa repentina o llanto) son comunes.
Los síntomas suelen progresar durante varios años antes de que se establezca una incapacidad total.
Atrofia muscular progresiva
La atrofia muscular progresiva puede aparecer a cualquier edad. Es similar a la esclerosis lateral amiotrófica, excepto en que los nervios motores del cerebro no se ven afectados. Además, esta enfermedad progresa más lentamente. No se produce espasticidad, pero los músculos se debilitan, se vuelven flácidos y se desgastan. Los síntomas iniciales son contracciones involuntarias o fasciculaciones de las fibras musculares.
Al principio se ven afectadas las manos, seguidas de los brazos, los hombros y las piernas. Finalmente afecta todo el cuerpo.
Muchas personas con atrofia muscular progresiva sobreviven 25 años o más.
Parálisis bulbar progresiva
En la parálisis bulbar progresiva se ven afectados los nervios que controlan los músculos de la masticación, la deglución y el habla, haciendo que estas funciones sean cada vez más difíciles. La voz tiene un tono nasal. En algunas personas, las emociones son cambiantes.
Debido a la dificultad para deglutir, frecuentemente se inhalan alimentos o saliva hacia los pulmones, lo que origina ahogos o atragantamientos y aumenta el riesgo de neumonía por aspiración.
La muerte, a menudo por neumonía, suele producirse entre 1 y 3 años después de que aparezcan los síntomas.
Diagnóstico de la ELA y otras enfermedades de las neuronas motoras
Evaluación médica
Pruebas como la resonancia magnética, la electromiografía y los estudios de conducción nerviosa
Análisis de sangre y a veces de orina y punción lumbar
Los médicos sospechan la existencia de una enfermedad de las neuronas motoras en los adultos que presentan debilidad muscular progresiva sin dolor ni pérdida de la sensibilidad. Los médicos preguntan a la persona afectada acerca de los siguientes aspectos:
Qué partes del cuerpo se ven afectadas
Cuándo comenzaron los síntomas
Qué síntomas aparecieron primero
Cómo se han modificado los síntomas con el tiempo
Esta información orienta al médico sobre la causa probable de los síntomas.
La debilidad muscular puede tener muchas causas. Para ayudar a reducir las posibilidades son necesarias las siguientes pruebas diagnósticas:
La electromiografía, en la que se registra la actividad eléctrica en los músculos, puede ayudar a determinar si el problema está en los nervios, en la unión neuromuscular o en los músculos.
También se realizan estudios de conducción nerviosa, que miden la rapidez con que los nervios transmiten los impulsos. En la enfermedad de las neuronas motoras, la velocidad de los impulsos no se ve afectada hasta fases tardías, de modo que si los impulsos son inesperadamente lentos en fases tempranas de la enfermedad, la causa de los síntomas puede ser otro trastorno.
Una resonancia magnética nuclear (RMN) del cerebro, y a veces de la médula espinal, para descartar alteraciones que causan síntomas similares.
Se realizan pruebas para determinar la existencia de otras enfermedades que puedan causar debilidad.
Para detectar otros trastornos, los médicos pueden hacer lo siguiente:
Análisis de sangre para detectar infecciones (como por ejemplo la sífilis) y trastornos metabólicos
Análisis de orina para detectar metales pesados (como plomo o mercurio) si la persona afectada ha estado expuesta a ellos
Una punción lumbar para detectar signos de inflamación en el líquido que rodea el encéfalo y la médula espinal (líquido cefalorraquídeo)
Pruebas genéticas para detectar trastornos hereditarios, como neuropatías hereditarias
Con el tiempo, las enfermedades de las neuronas motoras tienden a causar síntomas que son tan característicos que el diagnóstico es obvio sin ninguna prueba.
Tratamiento de la ELA y otras enfermedades de las neuronas motoras
Fisioterapia
Medicamentos para aliviar los síntomas
No existe tratamiento o cura específicos para las enfermedades de las neuronas motoras. Sin embargo, los investigadores continúan buscando tratamientos seguros y eficaces.
Los cuidados proporcionados por un equipo de diferentes tipos de profesionales de la salud (equipo multidisciplinario) ayudan a afrontar la invalidez progresiva. La fisioterapia ayuda a mantener la fuerza muscular y a mantener la flexibilidad en las articulaciones, con lo que contribuye a prevenir contracturas. El personal de enfermería u otros cuidadores deben dar de comer con cuidado a las personas con dificultad para tragar, con el fin de evitar atragantamientos. Algunas personas deben ser alimentadas mediante un tubo introducido en el estómago a través de la pared abdominal (tubo de gastrostomía).
Ciertos medicamentos pueden ayudar a aliviar los síntomas:
El baclofeno ayuda a que los músculos sean menos espásticos.El baclofeno ayuda a que los músculos sean menos espásticos.
La fenitoína y la quinina disminuyen los calambres.La fenitoína y la quinina disminuyen los calambres.
Los fármacos con efectos anticolinérgicos, como el glicopirrolato, se pueden utilizar para reducir el babeo porque uno de los efectos anticolinérgicos consiste en reducir la formación de saliva., como el glicopirrolato, se pueden utilizar para reducir el babeo porque uno de los efectos anticolinérgicos consiste en reducir la formación de saliva.
La amitriptilina y la fluvoxamina (antidepresivos) ayudan a las personas que sufren cambios emocionales o depresión. Un fármaco que combina dextrometorfano (un inhibidor de la tos) y quinidina puede controlar los cambios emocionales.La amitriptilina y la fluvoxamina (antidepresivos) ayudan a las personas que sufren cambios emocionales o depresión. Un fármaco que combina dextrometorfano (un inhibidor de la tos) y quinidina puede controlar los cambios emocionales.
En algunas personas con esclerosis lateral amiotrófica, el riluzol, un fármaco que protege las neuronas (neuroprotector), puede prolongar la vida durante unos meses. Se toma por vía oral. Edaravone, un nuevo fármaco, puede frenar hasta cierto punto la disminución de la funcionalidad en personas con esclerosis lateral amiotrófica. El fenilbutirato de sodio/taurursodiol parece retrasar la disminución de la capacidad funcional, al igual que tofersén, una En algunas personas con esclerosis lateral amiotrófica, el riluzol, un fármaco que protege las neuronas (neuroprotector), puede prolongar la vida durante unos meses. Se toma por vía oral. Edaravone, un nuevo fármaco, puede frenar hasta cierto punto la disminución de la funcionalidad en personas con esclerosis lateral amiotrófica. El fenilbutirato de sodio/taurursodiol parece retrasar la disminución de la capacidad funcional, al igual que tofersén, unaterapia antisentido, que también ralentiza la progresión de la enfermedad. Debe administrarse mediante inyección en una vena (vía intravenosa, IV), en un músculo (vía intramuscular, IM). El tofersen debe inyectarse en el espacio que rodea la médula espinal (inyección intratecal)., que también ralentiza la progresión de la enfermedad. Debe administrarse mediante inyección en una vena (vía intravenosa, IV), en un músculo (vía intramuscular, IM). El tofersen debe inyectarse en el espacio que rodea la médula espinal (inyección intratecal).
Si aparece dolor a medida que progresa la enfermedad (por ejemplo, al sentarse en una misma posición durante largo tiempo) se utilizan benzodiazepinas, que son sedantes suaves.
La cirugía para mejorar la deglución solo es eficaz para unas pocas personas con parálisis bulbar progresiva.
Dado que la esclerosis lateral amiotrófica y la parálisis bulbar progresiva son progresivas e incurables, se aconseja a las personas que padezcan alguna de estas enfermedades que especifiquen por anticipado el tipo de atención que desean recibir en los momentos finales de su vida.





