


Les tumeurs qui trouvent leur origine dans un os sont appelées tumeurs osseuses primitives. Les tumeurs osseuses primitives peuvent être non cancéreuses (bénignes) ou cancéreuses (malignes).
Quand un cancer est diagnostiqué, on procède à sa classification par stade. La détermination du stade permet d’évaluer le degré d’évolution d’un cancer, en utilisant comme critères le degré d’agressivité (sa probabilité de propagation, selon l’apparence des cellules tumorales observées au microscope), les dimensions de la tumeur, et sa propagation ou non dans les tissus adjacents ou les ganglions lymphatiques ou d’autres organes plus éloignés.
(Voir aussi Présentation des tumeurs osseuses et Présentation des cancers.)
Adamantinomes
Les adamantinomes sont des tumeurs rares qui se développent le plus souvent au niveau du tibia. Les tumeurs surviennent généralement chez les adolescents et les personnes de 20 ans, mais peuvent se développer à tout âge. Elles provoquent souvent une douleur et les personnes peuvent souvent sentir la tumeur sous la peau lorsqu’elles passent les doigts dessus.
Ces tumeurs se développent lentement et ont un bas grade de malignité. Cela signifie qu’elles sont moins à même de se propager (métastaser) que les autres tumeurs. Cependant, bien que cela soit rare, des métastases peuvent apparaître (principalement aux poumons).
Pour le diagnostic des adamantinomes, les médecins réalisent des radiographies et prélèvent un échantillon de tissu pour l’examiner au microscope (biopsie).
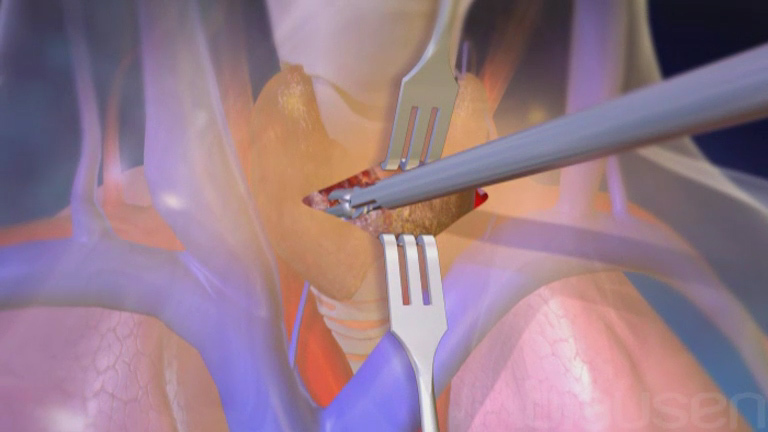
Pour traiter les adamantinomes, les médecins les enlèvent chirurgicalement sans inciser la tumeur, ce qui risquerait de propager les cellules tumorales. Si les cellules se propagent, le cancer peut récidiver. Dans de rares cas, l’ablation chirurgicale de la jambe touchée (amputation) peut s’avérer nécessaire en fonction de la localisation de la tumeur ou selon qu’il s’agisse d’une récidive.
Chondrosarcomes
Les chondrosarcomes sont des tumeurs composées de cellules cartilagineuses malignes. Ces tumeurs surviennent plutôt chez les adultes âgés. Ces tumeurs se développent souvent dans les os tels le bassin ou l’omoplate, mais peuvent se développer dans n’importe quelle partie de n’importe quel os et peuvent également se développer dans les tissus entourant les os. De nombreux chondrosarcomes sont des tumeurs à croissance lente ou à bas grade de malignité. Cela signifie qu’ils sont moins aptes à se propager (métastaser) que les autres tumeurs. Cependant, certains chondrosarcomes ont une croissance rapide ou un haut grade de malignité, et ont tendance à métastaser.
Pour diagnostiquer les chondrosarcomes, les médecins réalisent des radiographies, une scintigraphie osseuse et une imagerie par résonance magnétique (IRM). Les médecins prélèvent également un échantillon de tissu afin de l’examiner au microscope (biopsie).
Les chondrosarcomes à bas grade sont souvent retirés en grattant l’os avec un instrument semblable à une cuillère (curetage) et en utilisant de l’azote liquide, du phénol, un ciment osseux (méthacrylate de méthyle) ou un faisceau d’argon pour détruire les cellules tumorales superficielles incrustées dans l’os. Pratiquement toutes les tumeurs à bas grade sont guéries avec ces traitements chirurgicaux.
Les chondrosarcomes à grade élevé de malignité ou à croissance rapide sont des tumeurs agressives et sont plus à même de métastaser que certaines autres tumeurs. Ils doivent être retirés chirurgicalement en totalité sans couper la tumeur, ce qui risquerait de propager les cellules tumorales. Si les cellules se propagent, le cancer peut récidiver.
Les chondrosarcomes de tout grade ne répondent ni à la chimiothérapie ni à la radiothérapie. L’amputation du bras ou de la jambe affecté(e) est rarement nécessaire.
Chordomes
Les chordomes sont rares et cancéreux. Ils tendent à survenir aux extrémités de la colonne vertébrale, généralement au milieu de la base de la colonne (sacrum), au niveau du coccyx ou de la base du crâne. Un chordome affectant le sacrum ou le coccyx cause une douleur quasi permanente. Un chordome à la base du crâne peut causer des problèmes au niveau des nerfs à la base du crâne (nerfs crâniens). Les symptômes peuvent durer plusieurs mois ou plusieurs années avant que le diagnostic soit posé. En général, les chordomes ne se propagent (métastasent) pas aux autres régions, comme le poumon par exemple, sauf s’ils sont plus agressifs, mais ils peuvent récidiver après le traitement.
Pour faciliter le diagnostic des chordomes, les médecins peuvent réaliser une imagerie par résonance magnétique (IRM). Les médecins réalisent également une biopsie.
Les chordomes affectant le sacrum ou le coccyx peuvent être soignés par ablation chirurgicale. Les chordomes à la base du crâne ne peuvent généralement pas être soignés par intervention chirurgicale, mais la radiothérapie peut temporairement rétrécir la tumeur et soulager la douleur.
Sarcome osseux d’Ewing
Le sarcome d’Ewing est une tumeur maligne touchant plus souvent les hommes que les femmes, et plus fréquente chez les personnes âgées de 10 à 20 ans. La majorité de ces tumeurs se développent au niveau des bras ou des jambes, mais elles peuvent se développer dans n’importe quel os. La douleur et le gonflement sont les symptômes les plus fréquents. Les tumeurs peuvent devenir assez grosses et parfois affecter l’os sur toute sa longueur. La tumeur peut inclure une masse importante de tissus mous.
Pour diagnostiquer un sarcome d’Ewing, les médecins réalisent des radiographies. Même si les radiographies peuvent montrer des détails, l’imagerie par résonance magnétique (IRM) peut permettre de déterminer la taille exacte de la tumeur. Pour confirmer le diagnostic, les médecins réalisent une biopsie.


Image publiée avec l’aimable autorisation des Drs Michael J. Joyce et Hakan Ilaslan.
Le traitement du sarcome d’Ewing inclut différentes combinaisons (intervention chirurgicale, chimiothérapie et radiothérapie) selon que l’intervention chirurgicale est réalisable ou, si une tentative a été effectuée, si elle a donné de bons résultats. Ces associations thérapeutiques peuvent soigner plus de 60 % des personnes atteintes du sarcome d’Ewing.
Fibrosarcomes et sarcomes osseux indifférenciés pléomorphes
Les fibrosarcomes et les sarcomes osseux indifférenciés pléomorphes (anciennement appelés histiocytomes fibreux malins osseux) affectent des personnes du même groupe d’âge et sont similaires aux ostéosarcomes en termes d’aspect, de localisation et de symptômes. Ces tumeurs cancéreuses possèdent des cellules qui produisent des tissus fibreux (tissus conjonctifs) cancéreux et non des tissus osseux cancéreux.
Le traitement et les taux de survie sont similaires à ceux de l’ostéosarcome.
Lymphome osseux
Le lymphome osseux (anciennement appelé « sarcome à cellules réticulaires ») est une tumeur maligne touchant les personnes vers 40 ou 50 ans. Il peut débuter dans n’importe quel os ou dans n’importe quelle partie de l’organisme et se diffuser ensuite à la moelle osseuse. En général, cette tumeur cause des douleurs, un gonflement et une accumulation de tissus mous. L’os altéré a tendance à se fracturer.
Pour diagnostiquer un lymphome osseux, les médecins réalisent des radiographies et une imagerie par résonance magnétique (IRM). Une biopsie est également effectuée.
Le traitement du lymphome osseux consiste généralement en une chimiothérapie avec ou sans radiothérapie, ce qui semble être aussi efficace qu’une excision chirurgicale de la tumeur. L’amputation est rarement nécessaire. Si un os semble pouvoir se fracturer, les médecins peuvent le stabiliser chirurgicalement pour essayer de prévenir les fractures.
Tumeurs malignes à cellules géantes
Les tumeurs malignes à cellules géantes sont rares et cancéreuses et se trouvent généralement à l’extrémité d’un os long (os du bras ou de la cuisse). En général, ces tumeurs entraînent des douleurs et un gonflement.
Pour diagnostiquer une tumeur maligne à cellules géantes, les médecins réalisent des radiographies, ainsi qu’une imagerie par résonance magnétique (IRM) et une biopsie.
Le traitement de la tumeur maligne à cellules géantes est similaire à celui des ostéosarcomes, mais le taux de guérison est faible.
Myélome multiple
Le myélome multiple (voir aussi Maladies des plasmocytes : le myélome multiple) est parfois considéré comme un cancer du système hématologique (sanguin), mais il est aussi parfois considéré comme une tumeur osseuse. En tant que tumeur osseuse, il s’agit de la tumeur osseuse cancéreuse primitive (maligne) la plus fréquente. Elle survient principalement chez les adultes plus âgés. Cependant, ce cancer implique la moelle épinière (tissu formant le sang dans la cavité de l’os) et non dans les tissus durs qui constituent l’os. Ainsi, il est généralement plus considéré comme un cancer de la moelle osseuse que comme un cancer osseux. Il est plus fréquent que les cancers des tissus durs constituant l’os.
Les cellules médullaires cancéreuses sécrètent des substances entraînant la perte osseuse. Cette perte osseuse peut être diffuse ou, plus souvent, être restreinte à certaines zones au niveau des os.
Le myélome multiple peut affecter un ou plusieurs os, donc la douleur peut être localisée à un endroit ou plusieurs. Si un seul os est affecté par une seule tumeur, l’affection est appelée plasmacytome. S’il existe plusieurs tumeurs ou si la moelle osseuse est largement affectée, l’affection s’appelle myélome multiple.
Une biopsie osseuse est parfois effectuée pour poser le diagnostic dans les régions où l’os a été détruit. Si le myélome multiple est suggéré par les résultats de la biopsie osseuse ou que le myélome multiple est suspecté pour d’autres raisons, le diagnostic est confirmé par le prélèvement et l’examen de cellules de la moelle osseuse. Des analyses de sang sont également effectuées. En outre, les médecins prennent des radiographies de l’ensemble du corps (bilan osseux). L’imagerie par résonance magnétique (IRM) ou la tomographie par émission de positons (TEP) associée à la tomodensitométrie (TEP-TDM) peut aussi être réalisée pour examiner les sites spécifiques de douleur osseuse.


Image publiée avec l’aimable autorisation des Drs Michael J. Joyce et Hakan Ilaslan.
Le traitement du myélome multiple est complexe et peut inclure chimiothérapie, radiothérapie et parfois chirurgie.
Ostéosarcomes (sarcomes ostéogènes)
L’ostéosarcome est le type le plus fréquent de tumeur osseuse cancéreuse primitive si l’on considère le myélome multiple comme une tumeur hématologique. Même si les ostéosarcomes sont fréquents surtout chez les personnes âgées de 10 à 25 ans, ils peuvent survenir à n’importe quel âge. Il existe une prédisposition génétique, en particulier chez les enfants porteurs du gène du rétinoblastome héréditaire et du syndrome de Li-Fraumeni. Les personnes plus âgées atteintes de la maladie de Paget qui ont fait l’objet d’une radiothérapie osseuse pour un autre cancer, ou présentent des zones de tissu osseux mort (ce que l’on appelle infarctus osseux) et d’autres affections, développent parfois ce type de tumeur. Les ostéosarcomes se développent généralement dans ou autour du genou, mais peuvent apparaître dans n’importe quel os. Ils ont tendance à se diffuser (métastaser) dans les poumons ou d’autres os. En général, ces tumeurs entraînent des douleurs et un gonflement.
Des radiographies sont effectuées, mais le prélèvement d’un échantillon de tissu pour examen au microscope (biopsie) est nécessaire au diagnostic de l’ostéosarcome. Les personnes doivent passer une radiographie thoracique et une tomodensitométrie (TDM) pour détecter si le cancer s’est métastasé au poumon et une scintigraphie osseuse pour détecter si le cancer s’est propagé aux autres os. L’imagerie par résonance magnétique (IRM) et la tomographie par émission de positons (TEP) associée à la tomodensitométrie (TEP-TDM) sont d’autres examens d’imagerie également réalisés.


Image publiée avec l’aimable autorisation des Drs Michael J. Joyce et Hakan Ilaslan.
Plus de 65 % des personnes ayant ce type de tumeur survivent pendant au moins 5 ans après le diagnostic lorsqu’une chimiothérapie est administrée et que le cancer ne s’est pas métastasé. Si la chimiothérapie détruit pratiquement tout le cancer, la chance de survie d’au moins 5 ans est supérieure à 90 %. Dans la mesure où les interventions chirurgicales se sont améliorées, il est généralement possible de sauver le membre concerné et de le reconstruire. Par le passé, le membre touché devait souvent être amputé.
Les ostéosarcomes sont généralement traités en associant chimiothérapie et chirurgie. En général, le traitement commence par la chimiothérapie. La douleur persiste souvent pendant cette phase de traitement. Ensuite, la tumeur est retirée chirurgicalement sans la découper. Le fait de découper la tumeur propage ses cellules, ce qui peut entraîner une récidive du cancer dans la même région. La chimiothérapie continue après l’intervention chirurgicale.





