



- Overview of Glomerular Disorders
- Overview of Nephritic Syndrome
- Alport Syndrome
- Immunoglobulin A Nephropathy
- Postinfectious Glomerulonephritis (PIGN)
- Rapidly Progressive Glomerulonephritis (RPGN)
- Thin Basement Membrane Disease
- Overview of Nephrotic Syndrome
- Congenital Nephrotic Syndromes
- Diabetic Nephropathy
- Focal Segmental Glomerulosclerosis
- HIV-Associated Nephropathy
- Membranous Nephropathy
- Minimal Change Disease
- Fibrillary and Immunotactoid Glomerulopathies
- Lupus Nephritis
- Membranoproliferative Glomerulonephritis
Fibrillary glomerulopathy and immunotactoid glomerulopathy are rare conditions defined pathologically by organized deposition of nonamyloid microfibrillar or microtubular structures within the renal mesangium and basement membrane.

")
")
")
Fibrillary and immunotactoid glomerulopathies are found in approximately 0.6% of renal biopsy specimens. Average age at diagnosis is in the mid-50s with a slight female predominance for fibrillary glomerulopathy, which is the more common of the two. There is controversy about the relationship between the two; they are thought by some experts to be related disorders despite distinct clinical and pathologic features (1). Mechanism is unknown, although deposition of immunoglobulin, particularly IgG kappa and lambda light chains and complement (C3), suggests immune system dysfunction. Patients may have accompanying cancer, paraproteinemia, cryoglobulinemia, plasma cell dyscrasia, hepatitis C infection, or systemic lupus erythematous, or they may have a primary kidney disease without evidence of systemic disease. Immunotactoid glomerulopathy in particular is commonly associated with chronic lymphocytic leukemia and B cell lymphoma.
All patients have proteinuria, often the nephrotic range (≥ 3 g/day). Microscopic hematuria, hypertension, and chronic kidney disease may be detected at presentation.
(See also Overview of Nephrotic Syndrome.)
General reference
1. Rosenstock JL, Markowitz GS, Valeri AM, Sacchi G, Appel GB, D'Agati VD. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int 2003;63(4):1450-1461. doi:10.1046/j.1523-1755.2003.00853.x
Diagnosis of Fibrillary and Immunotactoid Glomerulopathies
Renal biopsy
Diagnosis is suggested by laboratory data and confirmed by renal biopsy. If nephrotic syndrome is present, testing is performed as for other cases of nephrotic syndrome.
Urinalysis usually shows features of nephritic syndrome and nephrotic syndrome.
Serum complement (C3 and C4) are usually measured and are occasionally decreased.
Light microscopy of a biopsy specimen shows mesangial expansion by amorphous eosinophilic deposits and mild mesangial hypercellularity. Various other changes may be present on light microscopy (eg, crescent formation, membranoproliferative patterns). Congo red staining is negative for amyloid. Immunostaining reveals IgG and C3 and sometimes kappa and lambda light chains in the area of the deposits.
Electron microscopy shows glomerular deposits consisting of extracellular, elongated, nonbranching microfibrils or microtubules. In fibrillary glomerulopathy, the diameter of the microfibrils and microtubules varies from 20 to 30 nm. In immunotactoid glomerulopathy, the diameter of the microfibrils and microtubules varies from 30 to 50 nm. In contrast, in amyloidosis, fibrils are 8 to 12 nm.

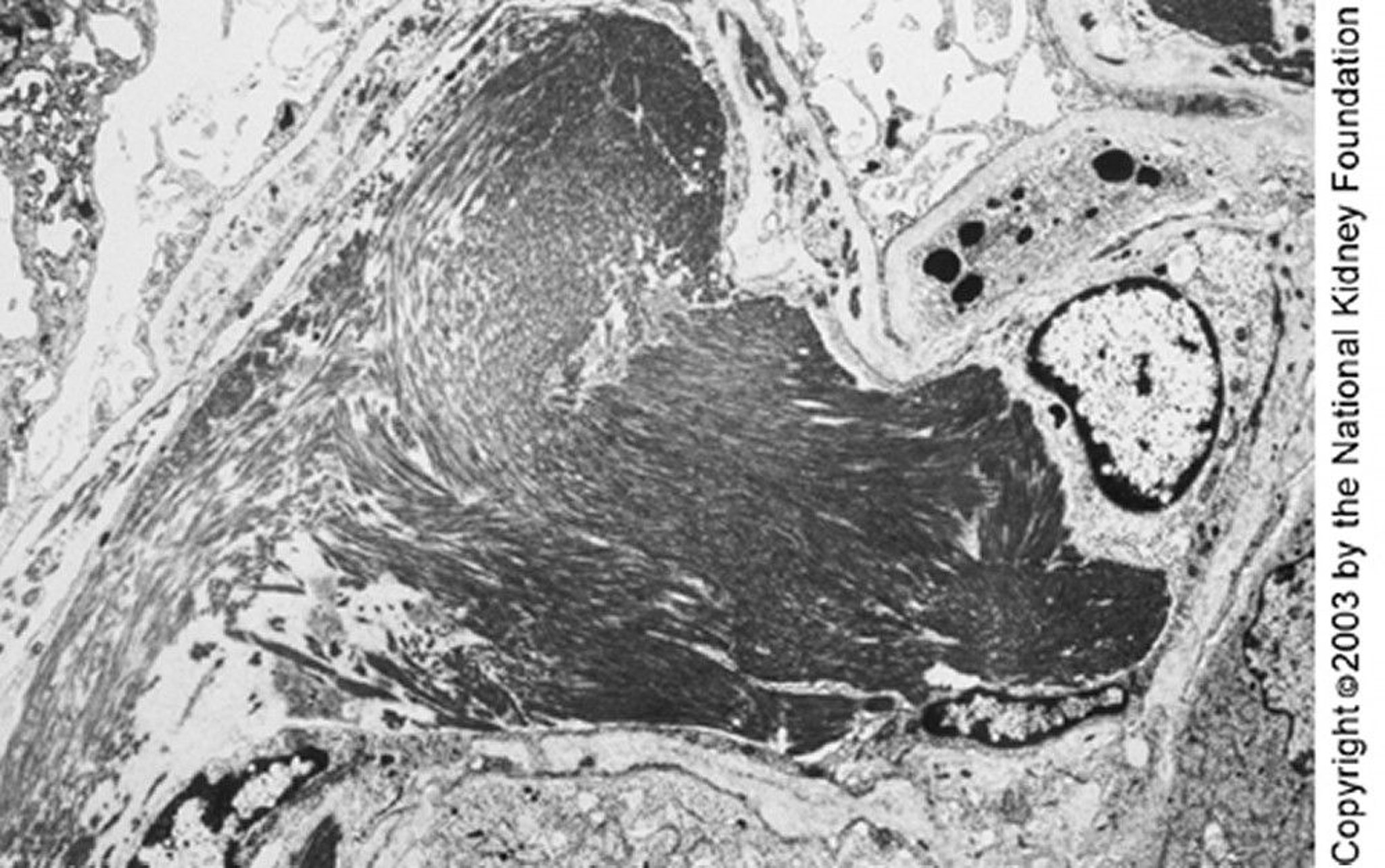
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Some experts distinguish immunotactoid from fibrillary glomerulopathy by the presence of microtubular (as opposed to smaller microfibrillar) structures in the deposits; others distinguish them by the presence of a related systemic illness. For example, a lymphoproliferative disorder, monoclonal gammopathy, cryoglobulinemia, or systemic lupus erythematosus may suggest immunotactoid glomerulopathy.

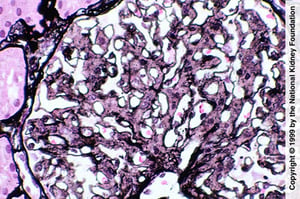
Mesangial proliferation is suggestive of fibrillary glomerulopathy; however, the diagnosis requires negative Congo red stain, IgG staining by immunofluorescence, and demonstration of fibril on electron microscopy (Jones silver stain, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
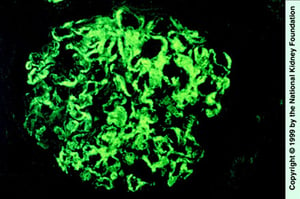
Smudgy IgG deposition in mesangium and capillary loops (immunofluorescence staining with anti-IgG, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

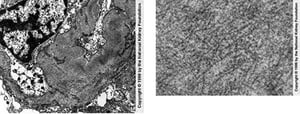
Randomly arranged fibrils in mesangial and capillary loops are seen on transmission electron microscopy (left). Negative Congo red stains are necessary to exclude renal amyloidosis (×25,625). The right image shows a high-power view of fibrils, which are coarser in diameter than amyloid deposits (×98,000).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Mesangial proliferation is suggestive of fibrillary glomerulopathy; however, the diagnosis requires negative Congo red stain, IgG staining by immunofluorescence, and demonstration of fibril on electron microscopy (Jones silver stain, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Smudgy IgG deposition in mesangium and capillary loops (immunofluorescence staining with anti-IgG, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Randomly arranged fibrils in mesangial and capillary loops are seen on transmission electron microscopy (left). Negative Congo red stains are necessary to exclude renal amyloidosis (×25,625). The right image shows a high-power view of fibrils, which are coarser in diameter than amyloid deposits (×98,000).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Treatment of Fibrillary and Immunotactoid Glomerulopathies
Sometimes, rituximabSometimes, rituximab
Consider angiotensin-converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs), corticosteroids and/or other immunosuppressants
The underlying condition, if found, should be treated.
Some evidence supports the use of rituximab in both fibrillary and immunotactoid glomerulopathies (Some evidence supports the use of rituximab in both fibrillary and immunotactoid glomerulopathies (1, 2). ACE inhibitors and ARBs may be used to reduce proteinuria. Corticosteroids have also been used. The disorder may recur after transplantation.
Treatment references
1. Marinaki S, Tsiakas S, Liapis G, et al. Clinicopathologic features and treatment outcomes of patients with fibrillary glomerulonephritis: A case series. Medicine (Baltimore) 2021;100(20):e26022. doi:10.1097/MD.0000000000026022
2. Javaugue V, Dufour-Nourigat L, Desport E, et al. Results of a nation-wide cohort study suggest favorable long-term outcomes of clone-targeted chemotherapy in immunotactoid glomerulopathy. Kidney Int 2021;99(2):421-430. doi:10.1016/j.kint.2020.06.039
Prognosis for Fibrillary and Immunotactoid Glomerulopathies
Fibrillary glomerulopathy progresses to kidney failure in 50% of patients within 4 years, with likely more favorable outcomes in treated immunotactoid glomerulopathy (1, 2). A more rapid decline may be predicted by the presence of hypertension, nephrotic-range proteinuria, and chronic kidney disease at presentation.
Prognosis references
1. Javaugue V, Dufour-Nourigat L, Desport E, et al. Results of a nation-wide cohort study suggest favorable long-term outcomes of clone-targeted chemotherapy in immunotactoid glomerulopathy. Kidney Int 2021;99(2):421-430. doi:10.1016/j.kint.2020.06.039
2. Nasr SH, Valeri AM, Cornell LD, et al. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol 2011;6(4):775-784. doi:10.2215/CJN.08300910







