



- Overview of Glomerular Disorders
- Overview of Nephritic Syndrome
- Alport Syndrome
- Immunoglobulin A Nephropathy
- Postinfectious Glomerulonephritis (PIGN)
- Rapidly Progressive Glomerulonephritis (RPGN)
- Thin Basement Membrane Disease
- Overview of Nephrotic Syndrome
- Congenital Nephrotic Syndromes
- Diabetic Nephropathy
- Focal Segmental Glomerulosclerosis
- HIV-Associated Nephropathy
- Membranous Nephropathy
- Minimal Change Disease
- Fibrillary and Immunotactoid Glomerulopathies
- Lupus Nephritis
- Membranoproliferative Glomerulonephritis
Alport syndrome is a genetically heterogeneous disorder characterized by nephritic syndrome (ie, hematuria, proteinuria, hypertension, eventual renal insufficiency) often with sensorineural deafness and, less commonly, ophthalmologic symptoms. Cause is a gene mutation affecting type IV collagen. Diagnosis is by history, including family history, urinalysis, and biopsy (renal or skin). Treatment is the same as that for chronic kidney disease, sometimes including kidney transplantation.
Topic Resources
")
(See also Overview of Nephritic Syndrome.)
Alport syndrome is a nephritic syndrome caused by a mutation in the COL4A3, COL4A4, and COL4A5 genes that encode the alpha-5 chain of type IV collagen and results in altered type IV collagen strands. The mechanism by which collagen alteration causes a glomerular disorder is unknown, but impaired structure and function are presumed; in most families, thickening and thinning of the glomerular and tubular basement membranes occur, with multilamination of the lamina densa in a focal or local distribution (basket-weave pattern). Glomerular scarring and interstitial fibrosis eventually result.

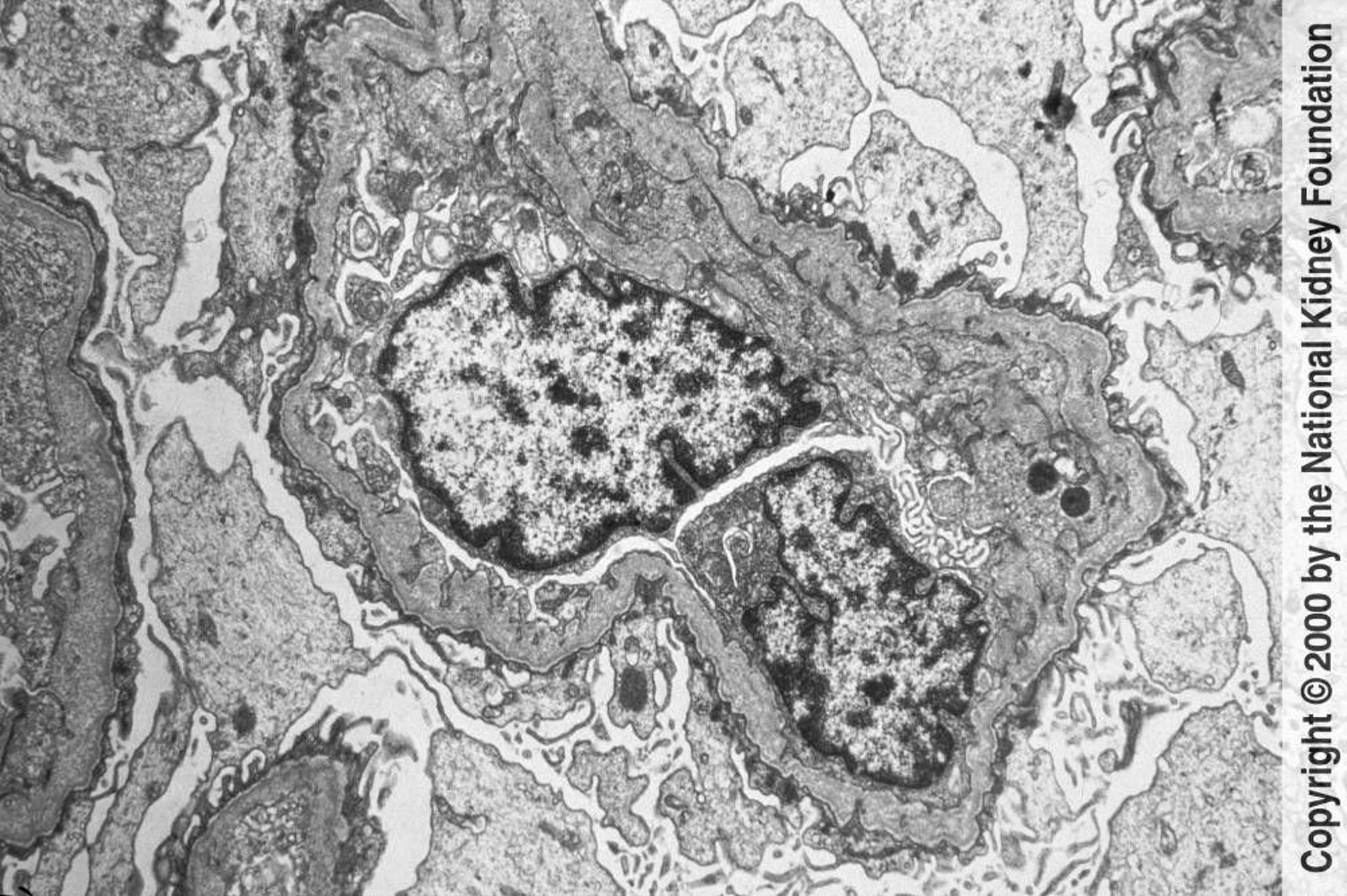
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
The disorder is most commonly inherited in X-linked fashion, although autosomal recessive and, rarely, autosomal dominant varieties exist. Cases with X-linked inheritance may be clinically categorized as
Juvenile form: Renal insufficiency develops between age 20 and 30 years
Adult form: Renal insufficiency develops in people > 30 years
Symptoms and Signs of Alport Syndrome
Classic X-linked disease in males and autosomal recessive disease are clinically similar. Patients develop renal symptoms and signs similar to those of acute nephritic syndrome (eg, microscopic hematuria, hypertension, eventually gross hematuria with proteinuria) and progress to renal insufficiency between ages 20 and 30 (juvenile forms).
Sensorineural hearing loss frequently is present, affecting higher frequencies; it may not be noticed during early childhood.
Ophthalmologic abnormalities—cataracts (most common), anterior lenticonus (a regular conical protrusion on the anterior aspect of the lens due to thinning of the lens capsule), spherophakia (spherical lens deformation that can predispose to lens subluxation), nystagmus, retinitis pigmentosa, blindness—also occur but less frequently than hearing loss.
X-linked disease occurs in heterozygous women, who, because they have one normal X chromosome, usually have less severe, more slowly progressing symptoms than men.
Some men with X-linked disease develop renal insufficiency after age 30 with hearing loss that occurs late or is mild, and autosomal dominant disease typically does not cause renal failure until age ≥ 45 years (adult forms).
In some patients with X-linked disease, sensorineural hearing loss usually manifests in childhood, whereas renal disease often does not manifest until adulthood.
Other nonrenal manifestations rarely include polyneuropathy and thrombocytopenia.
Diagnosis of Alport Syndrome
Serum creatinine level
Urinalysis
Renal biopsy
Molecular genetic analysis
Diagnosis is suggested in patients who have microscopic hematuria on urinalysis or recurrent episodes of gross hematuria, particularly if an abnormality of hearing or vision or a family history of chronic kidney disease is present.
Serum creatinine level is checked to assess renal function. Urinalysis and usually renal biopsy are also done. In addition to dysmorphic red blood cells, the urine may contain protein, white blood cells, and casts of various types. Nephrotic syndrome occurs rarely. No distinguishing histologic changes are seen on light microscopy. The diagnosis can be confirmed by any of the following:
Renal biopsy with immunostaining for the subtypes of type IV collagen
Characteristic disorganization of the lamina densa with variable thickening and thinning of the glomerular capillary basement membrane seen using electron microscopy
Skin biopsy with immunostaining for the type IV collagen subtypes in a patient with a positive family history
Molecular genetic analysis of the COL4A genes
A combination of immunostaining and electron microscopy is often needed to distinguish Alport syndrome from some forms of thin basement membrane disease.
Treatment of Alport Syndrome
Same as that for other causes of chronic kidney disease
Kidney transplantation
Early renin-angiotensin blockade with an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin II receptor blocker (ARB) has been shown to slow the progression of disease (Early renin-angiotensin blockade with an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin II receptor blocker (ARB) has been shown to slow the progression of disease (1). Its management is similar to that for other causes of chronic kidney disease. Transplantation has been successful, but antiglomerular basement membrane antibody disease may occur, usually only in males, in the transplanted kidney. Genetic counseling is indicated.
Hearing loss and/or ocular impairment are managed with supportive measures (eg, hearing aids).
Treatment reference
1. Gross O, Tönshoff B, Weber LT, et al: A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport's syndrome. Kidney Int 97(6):1275-1286, 2020. doi: 10.1016/j.kint.2019.12.015
Key Points
Consider Alport syndrome if patients have hematuria plus a hearing and/or vision abnormality or a family history of chronic kidney disease.
Confirm the diagnosis by biopsy of the kidney or sometimes skin and immunostaining for type IV collagen subtypes or molecular genetic testing.
Treat chronic kidney disease and consider transplantation.







