


Down syndrome is an abnormality of chromosome 21 that can cause intellectual disability, microcephaly, short stature, and characteristic facies. Diagnosis is suggested by physical anomalies and abnormal development and confirmed by cytogenetic analysis. Management depends on specific manifestations and anomalies.
(See also Overview of Chromosomal Abnormalities.)
Overall incidence in the United States is approximately 1/700 live births (1), and the risk increases gradually with increasing maternal age. Based on a large study, at 20 years of maternal age, the risk is 1/1466 births; at 35, it is 1/343; and at 40, it is 1/85 (2). However, because most births occur among younger women, the majority of children with Down syndrome are born to women < 35 years; only approximately 20% of infants with Down syndrome are born to mothers > 35 years.
References
1. Mai CT, Isenburg JL, Canfield MA, et al: National population-based estimates for major birth defects, 2010-2014. Birth Defects Res 111(18):1420-1435, 2019. doi: 10.1002/bdr2.1589
2. Morris JK, Mutton DE, Alberman E: Revised estimates of the maternal age specific live birth prevalence of Down's syndrome. J Med Screen 9(1):2-6, 2002. doi: 10.1136/jms.9.1.2
Etiology of Down Syndrome
In approximately 95% of cases, Down syndrome is caused by nondisjunction resulting in an extra chromosome 21 (trisomy 21), which is typically maternally derived (1). Such people have 47 chromosomes instead of the normal 46.

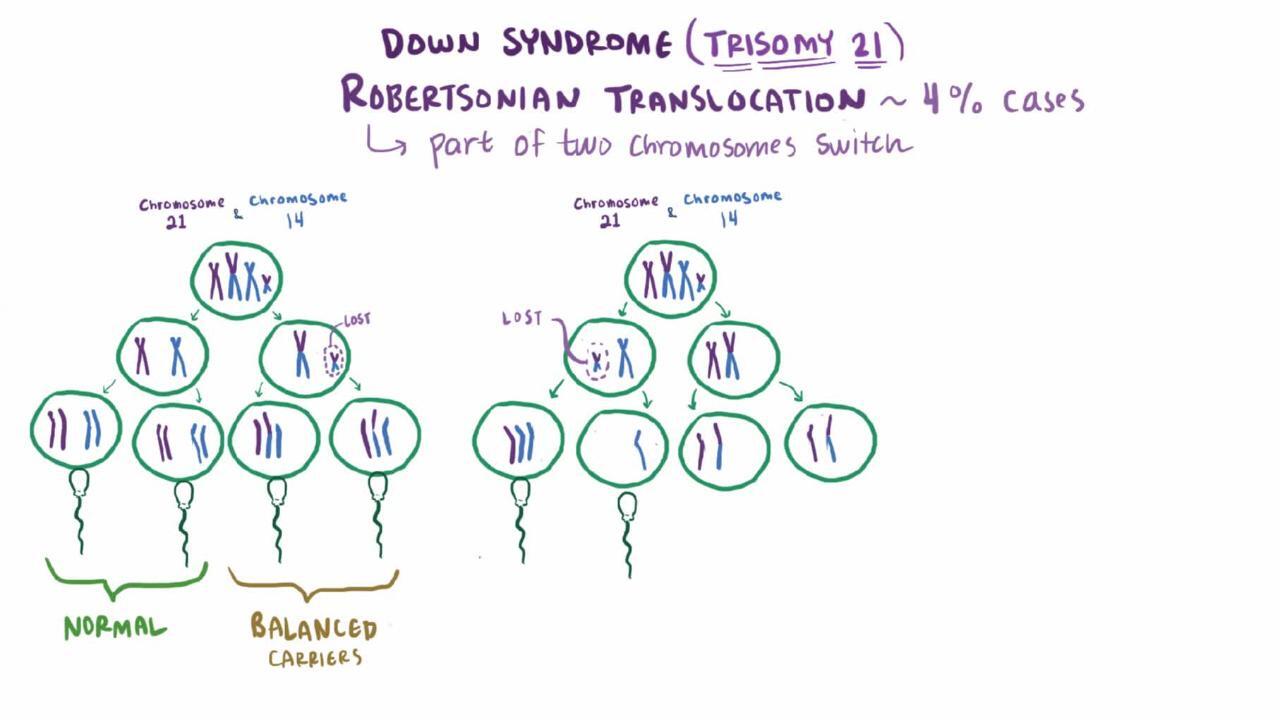
Approximately 4% of Down syndrome cases are due to a translocation (2). In a balanced translocation, genetic material is exchanged with material from another nonhomologous chromosome, and the chromosome count is maintained at 46.
The most common translocation is t(14;21), in which chromosome 21 is attached to chromosome 14; this is an unbalanced translocation, resulting in a chromosome count of 45. In about half of people with the t(14;21) translocation, both parents have normal karyotypes, indicating a de novo translocation. In the other half, one parent (almost always the mother), who does not have Down syndrome, has only 45 chromosomes, one of which is t(14;21). Theoretically, the chance that a carrier mother will have a child with Down syndrome is 1:3, but the actual risk is lower (approximately 1:10). If the father is the carrier, the risk is only 1:20.
The next most common translocation is t(21;22). In these cases, carrier mothers have approximately a 1:10 risk of having a child with Down syndrome; the risk is smaller for carrier fathers.
Translocation 21q;21q, which occurs when the extra chromosome 21 is attached to another chromosome 21, is much less common. It is particularly important to determine whether a parent is a carrier of, or a mosaic for, translocation 21q;21q. Each child of a carrier of the translocation will have Down syndrome or monosomy 21. Because monosomy 21 is not typically compatible with life, the risk of having a viable child with Down syndrome is 100%. If the parent is mosaic, that parent has some normal cells and some 45-chromosome cells with 21q;21q, and so the risk of Down syndrome is markedly increased, although these people may also have children with normal chromosomes.
Down syndrome mosaicism presumably results from nondisjunction (when chromosomes fail to pass to separate cells) during cell division in the embryo. People with mosaic Down syndrome have two cell lines, one with the normal 46 chromosomes and another with 47 chromosomes, including an extra chromosome 21. The prognosis for intelligence and risk of medical complications probably depends on the proportion of trisomy 21 cells in each different tissue, including the brain. However, in practice, risk cannot be predicted because it is not feasible to determine the karyotype in every single cell in the body. Some people with mosaic Down syndrome have very subtle clinical signs and may have normal intelligence; however, even people with no detectable mosaicism can have very variable findings. If a parent has germline mosaicism for trisomy 21, an increased risk, above the maternal age-based risk, exists for a second affected child.
Etiology references
1. Antonarakis SE: Parental origin of the extra chromosome in trisomy 21 as indicated by analysis of DNA polymorphisms. Down Syndrome Collaborative Group. N Engl J Med 324(13):872-876, 1991. doi: 10.1056/NEJM199103283241302
2. Mutton D, Alberman E, Hook EB: Cytogenetic and epidemiological findings in Down syndrome, England and Wales 1989 to 1993. National Down Syndrome Cytogenetic Register and the Association of Clinical Cytogeneticists. J Med Genet 33(5):387-394, 1996. doi: 10.1136/jmg.33.5.387
Pathophysiology of Down Syndrome
As with most conditions that result from chromosome imbalance, Down syndrome affects multiple systems and causes both structural and functional defects (see table Some Complications of Down Syndrome). Not all defects are present in each person.
Some Complications of Down Syndrome*
System | Deficit |
|---|---|
Cardiac | Congenital heart disease, most often ventricular septal defect and atrioventricular canal defect Increased risk of mitral valve prolapse and aortic regurgitation (more frequently seen in adults) |
Central nervous system | Cognitive impairment (mild to severe) Motor and language delay |
Gastrointestinal | |
Endocrine | |
Eyes, ears, nose, and throat | Ophthalmic disorders (eg, congenital cataracts, glaucoma, strabismus, refractive errors) Increased incidence of otitis media |
Growth | Short stature |
Hematologic | Thrombocytopenia Transient myelodysplastic disorder Acute megakaryoblastic leukemia |
Musculoskeletal | Atlantoaxial and atlanto-occipital instability Joint laxity |
Respiratory | |
* Not all complications are present in a given patient, but incidence is increased compared with unaffected population. | |
Most affected people have some degree of cognitive impairment, ranging from severe (IQ 20 to 35) to mild (IQ 50 to 75). Gross motor and language delays also are evident early in life. Height is often reduced, and there is an increased risk of obesity.
Approximately 50% of affected neonates have congenital heart disease; ventricular septal defect and atrioventricular canal defect (endocardial cushion defect) are most common (1, 2).
Approximately 6% of affected people have gastrointestinal anomalies, particularly duodenal atresia, sometimes along with annular pancreas. Hirschsprung disease and celiac disease also are more common (3).
Many people develop endocrinopathies, including thyroid disease (most often hypothyroidism) and diabetes.
Atlanto-occipital and atlantoaxial hypermobility, as well as bony anomalies of the cervical spine, can cause atlanto-occipital and cervical instability; weakness and paralysis may result.
Approximately 60% of people have eye problems, including congenital cataracts, glaucoma, strabismus, and refractive errors.
Most people have hearing loss, and ear infections are very common.
The aging process seems to be accelerated (4). In recent decades, the median life expectancy has increased to about 60 years, and some affected people live into their 80s (5). Comorbidities contributing to decreased life expectancy include heart disease, increased susceptibility to infections, and leukemia. There is an increased risk of Alzheimer disease at an early age, and, at autopsy, brains of adults with Down syndrome show typical microscopic findings (6). Research indicates that Black people with Down syndrome have a substantially shorter life span than White people (7, 8). This finding may be the result of poor access to medical, educational, and other support services.
Affected women have a 50% chance of having a fetus that also has Down syndrome; however, there appears to be an increased risk of spontaneous abortion. Men with Down syndrome are infertile, except for those with mosaicism.
Pathophysiology references
1. Irving CA, Chaudhari MP: Cardiovascular abnormalities in Down's syndrome: Spectrum, management and survival over 22 years. Arch Dis Child 97(4):326-330, 2012. doi: 10.1136/adc.2010.210534
2. de Groot-van der Mooren MD, Scheerman BC, Rammeloo LAJ, et al: Neonatal mortality and morbidity in Down syndrome in the time of prenatal aneuploidy testing: A retrospective cohort study. Eur J Pediatr 182(1):319-328, 2023. doi: 10.1007/s00431-022-04686-3
3. Stoll C, Dott B, Alembik Y, Roth MP: Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet 58(12):674-680, 2015. doi: 10.1016/j.ejmg.2015.11.003
4. Horvath S, Garagnani P, Bacalini MG, et al: Accelerated epigenetic aging in Down syndrome. Aging Cell 14(3):491-495, 2015. doi: 10.1111/acel.12325
5. Englund A, Jonsson B, Zander CS, et al: Changes in mortality and causes of death in the Swedish Down syndrome population. Am J Med Genet A 161A(4):642-649, 2013. doi: 10.1002/ajmg.a.35706
6. Davidson YS, Robinson A, Prasher VP, Mann DMA: The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer's disease in individuals with Down syndrome. Acta Neuropathol Commun 6(1):56, 2018. doi: 10.1186/s40478-018-0559-4
7. Kucik JE, Shin M, Siffel C, Marengo L, Correa A; Congenital Anomaly Multistate Prevalence and Survival Collaborative: Trends in survival among children with Down syndrome in 10 regions of the United States. Pediatrics 131(1):e27-e36, 2013. doi: 10.1542/peds.2012-1616
8. Santoro SL, Esbensen AJ, Hopkin RJ, et al: Contributions to Racial Disparity in Mortality among Children with Down Syndrome. J Pediatr 174:240-246.e1, 2016. doi: 10.1016/j.jpeds.2016.03.023
Symptoms and Signs of Down Syndrome
General appearance
Affected neonates tend to be placid, rarely cry, and have hypotonia. Most have a flat facial profile (particularly flattening of the bridge of the nose), but some do not have obviously unusual physical characteristics at birth and then develop more noticeable characteristic facial features during infancy. A flattened occiput, microcephaly, and extra skin around the back of the neck are common.
Typical physical features of people with Down syndrome include the following:
Eyes have an upward angle at the lateral edge, epicanthal folds at the inner corners usually are present, and Brushfield spots (gray to white spots resembling grains of salt around the periphery of the iris) may be visible.
Mouth is often held open; a protruding, furrowed tongue may lack the central fissure.
Ears are often small and rounded.
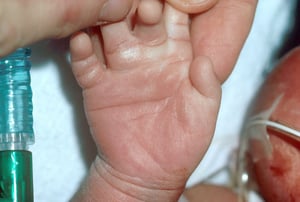
Hands are often short and broad and often have a single transverse palmar crease, and fingers are often short, with clinodactyly (incurving) of the 5th digit, which often has only 2 phalanges.
Feet may have a wide gap between the 1st and 2nd toes (sandal-gap toes), and a plantar furrow often extends backward on the foot.

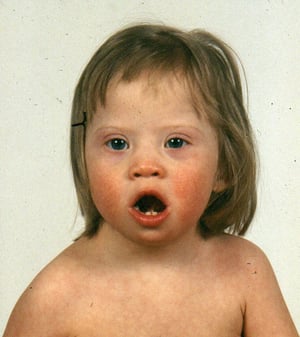
This photo shows a child with Down syndrome with a flattened nasal bridge, upslanting eyes, and epicanthal folds at the inner corners of the eyes.
© Springer Science+Business Media

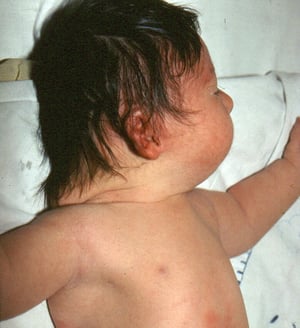
This photo shows redundant nuchal folds in a baby with Down syndrome.
© Springer Science+Business Media

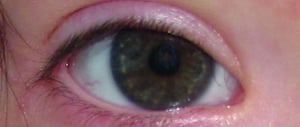
This photo shows white spots on the iris of a patient with Down syndrome.
© Springer Science+Business Media

RALPH C. EAGLE, JR./SCIENCE PHOTO LIBRARY


This photo shows a young man with many typical physical characteristics of Down syndrome such as short stature, frontal balding, thin hair, epicanthal folds, thick neck, and mild truncal obesity.
By permission of the publisher. From Bird T, Sumi S: Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002.
This photo shows a child with Down syndrome with a flattened nasal bridge, upslanting eyes, and epicanthal folds at the inner corners of the eyes.
© Springer Science+Business Media
This photo shows redundant nuchal folds in a baby with Down syndrome.
© Springer Science+Business Media
This photo shows white spots on the iris of a patient with Down syndrome.
© Springer Science+Business Media
RALPH C. EAGLE, JR./SCIENCE PHOTO LIBRARY
This photo shows a young man with many typical physical characteristics of Down syndrome such as short stature, frontal balding, thin hair, epicanthal folds, thick neck, and mild truncal obesity.
By permission of the publisher. From Bird T, Sumi S: Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002.
Growth and development
As affected children grow, delay of physical and mental development becomes apparent. Stature is often short. The mean IQ is about 50, but this varies widely. Behavior suggestive of attention-deficit/hyperactivity disorder is often present in childhood, and the incidence of autistic behavior is increased (particularly in children with profound intellectual disability).
There is an increased risk of depression in children and adults with Down syndrome.
Cardiac manifestations
Symptoms of heart disease are determined by the type and extent of the cardiac anomaly.
Infants with congenital heart defects, the most common of which are ventricular septal defects and atrioventricular canal defects, can either be asymptomatic or show signs of heart failure (eg, labored breathing, fast respiratory rate, difficulty with feeding, sweating, poor weight gain).
Murmurs may not be present; however, a number of different murmurs are possible.
Gastrointestinal manifestations
Infants with Hirschsprung disease usually have delay in passage of meconium. Severely affected infants may have signs of intestinal obstruction (eg, bilious vomiting, failure to pass stool, abdominal distention).
Duodenal atresia or stenosis can manifest with bilious vomiting or with no symptoms, depending on the extent of the stenosis. These defects may be detected by prenatal ultrasonography (double-bubble sign).


Diagnosis of Down Syndrome
Prenatal chorionic villus sampling and/or amniocentesis with karyotyping
Postnatal karyotyping (if prenatal karyotyping not done)
(See also Next-generation sequencing technologies.)
Diagnosis of Down syndrome may be suspected prenatally based on
Physical anomalies detected by fetal ultrasonography
Maternal serum screening
Noninvasive prenatal screening (NIPS)
Fetal ultrasonographic anomalies include increased nuchal translucency, atrioventricular canal defect, and duodenal atresia, but these are not present in all fetuses with trisomy 21.
Maternal serum testing may show abnormal levels of plasma protein A in late first trimester and alpha-fetoprotein, beta-hCG (human chorionic gonadotropin), unconjugated estriol, and inhibin in early second trimester (15 to 16 weeks gestation). Maternal serum testing may show abnormal levels of plasma protein A in late first trimester and alpha-fetoprotein, beta-hCG (human chorionic gonadotropin), unconjugated estriol, and inhibin in early second trimester (15 to 16 weeks gestation).
More recently, noninvasive prenatal screening (NIPS), in which fetal DNA obtained from the maternal circulation is tested, has become a screening option for trisomy 21 because it has good sensitivity and specificity.
If Down syndrome is suspected based on maternal serum screening tests or ultrasonography, fetal or postnatal confirmatory testing is recommended. Fetal confirmatory methods include chorionic villus sampling and/or amniocentesis with testing by karyotype analysis. Karyotyping is the test of choice to rule out an associated translocation so that parents can receive appropriate genetic counseling regarding recurrence risk. The option of prenatal confirmatory testing is offered to all patients with an abnormal, indeterminate, or unclear NIPS result. Management decisions, including termination of pregnancy, should not be made based on NIPS alone.
In high-resource settings, maternal serum screening and diagnostic testing for Down syndrome are usually available to all women who present for prenatal care before 20 weeks gestation regardless of maternal age.
The American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Obstetrics, Committee of Genetics, and the Society for Maternal–Fetal Medicine 2020 practice bulletin regarding screening for chromosomal abnormalities advises that cell-free fetal DNA analysis be offered to all pregnant women regardless of age or additional risk factors.
If diagnosis is not made prenatally, then neonatal diagnosis is based on physical anomalies and confirmed by cytogenetic testing, preferably karyotyping.
Concomitant medical conditions
Certain age-specific routine screening of affected infants and children helps identify conditions associated with Down syndrome (1):
Echocardiography: At prenatal visit or at birth
Thyroid screening (thyroid-stimulating hormone [TSH] levels): At birth, 6 months, 12 months, and annually thereafter
Complete blood count (CBC) with differential and either a combination of ferritin and C-reactive protein (CRP) or a combination of serum iron and total iron-binding capacity (TIBC): At 1 year and annually thereafter
Hearing evaluations: At birth, every 6 months thereafter until normal hearing established (about age 4 years), then annually (more frequently if indicated)
Ophthalmology evaluation: By 6 months, then annually until age 5; then every 2 years until age 13 and every 3 years until age 21 (more frequently if indicated)
Growth: Height, weight, and head circumference plotted at each health supervision visit using a Down syndrome growth chart
Sleep study for obstructive sleep apnea: Completed by age 4 years
Routine screening for atlantoaxial instability and celiac disease is not recommended; children are tested based on clinical suspicion. It is recommended that patients with a history of neck pain, radicular pain, weakness, or any other neurologic symptoms that suggest myelopathy have x-rays of the cervical spine in the neutral position; if no suspicious abnormalities are seen, they should have x-rays done in flexion and extension positions.
Medical care guidelines for adults with Down syndrome published by a panel of experts includes recommendations to screen for the following associated diseases (with recommended age to begin screening) (2):
Diabetes: Hemoglobin A1C or fasting glucose every 2 to 3 years starting at age 30 or at age 21 for those with comorbid obesity
Hypothyroidism: Thyroid-stimulating hormone (TSH) every 1 to 2 years starting at age 21
Alzheimer-type dementia: Annual assessment starting at age 40
Diagnosis references
1. Bull MJ, Trotter T, Santoro SL, et al: Health supervision for children and adolescents with Down syndrome. Pediatrics 149(5):e2022057010, 2022. doi: 10.1542/peds.2022-057010
2. Tsou AY, Bulova P, Capone G, et al: Medical Care of Adults With Down Syndrome: A Clinical Guideline. JAMA 324(15):1543-1556, 2020. doi: 10.1001/jama.2020.17024
Treatment of Down Syndrome
Screening for complications and associated diseases
Treatment of specific manifestations
Genetic counseling
The underlying genetic abnormality cannot be cured. Management depends on specific manifestations, and surveillance for complications or associated diseases is fairly uniform for all children (see Concomitant medical conditions).
Some congenital cardiac or gastrointestinal anomalies are repaired surgically. Other diseases (eg, hypothyroidism, celiac disease, leukemia) are treated as appropriate.
Care of children with Down syndrome and their families should also include genetic counseling for the family, social support, and educational programming appropriate for the level of intellectual functioning (see Intellectual Disability).
Key Points
Down syndrome involves an extra chromosome 21, either a separate chromosome or a translocation onto another chromosome.
Diagnosis may be suspected prenatally based on anomalies detected by fetal ultrasonography (eg, increased nuchal translucency, heart defect, duodenal atresia) or based on cell-free fetal DNA analysis of maternal blood or maternal multiple marker screening for levels of plasma protein A in late first trimester and levels of alpha-fetoprotein, beta-human chorionic gonadotropin (beta-hCG), unconjugated estriol, and inhibin in early second trimester. Diagnosis may be suspected prenatally based on anomalies detected by fetal ultrasonography (eg, increased nuchal translucency, heart defect, duodenal atresia) or based on cell-free fetal DNA analysis of maternal blood or maternal multiple marker screening for levels of plasma protein A in late first trimester and levels of alpha-fetoprotein, beta-human chorionic gonadotropin (beta-hCG), unconjugated estriol, and inhibin in early second trimester.
Karyotype analysis is the confirmatory test of choice and can be done prenatally by chorionic villus sampling in the first trimester or amniocentesis in the second trimester, or postnatally on a blood sample.
Life expectancy is decreased primarily because of heart disease and, to a lesser degree, increased susceptibility to infections, acute myelocytic leukemia, and early-onset Alzheimer disease; however, it has increased remarkably in recent decades, and some affected people live into their 80s.
Do routine age-specific screening to detect associated medical conditions (eg, cardiac anomalies, hypothyroidism).
Treat specific manifestations, and provide social and educational support and genetic counseling.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Obstetrics, Committee of Genetics, and the Society for Maternal–Fetal Medicine: Screening for fetal chromosomal abnormalities: ACOG practice bulletin, number 226 (2020)
American Academy of Pediatrics: Health supervision for children and adolescents with Down syndrome (2022)







